Bai Shuming, Mansour Ritam, Stojanović Ljiljana, Toldo Josene M, Barbatti Mario
Aix Marseille University, CNRS, ICR, Marseille, France.
Department of Chemistry, Duke University, Durham, NC, 27708, USA.
J Mol Model. 2020 Apr 21;26(5):107. doi: 10.1007/s00894-020-04355-y.
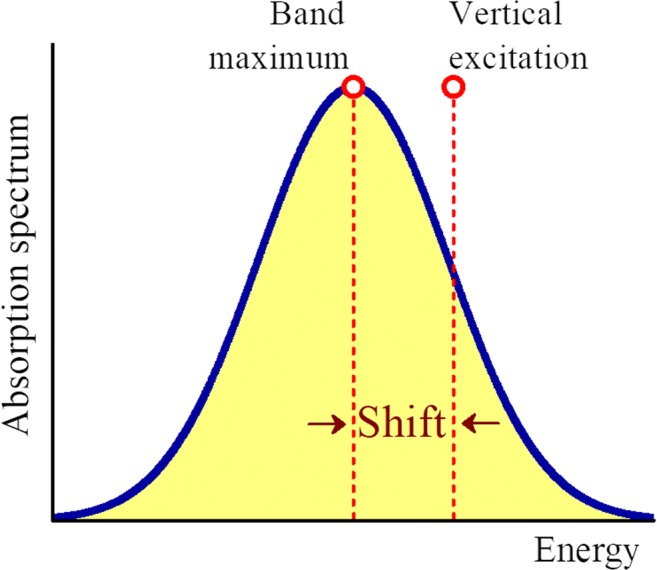
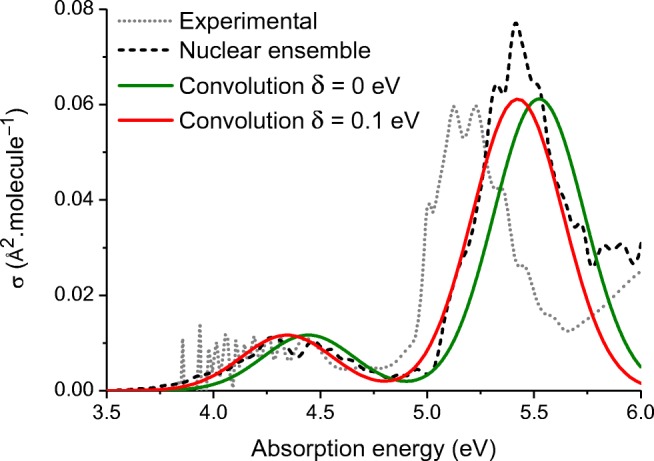
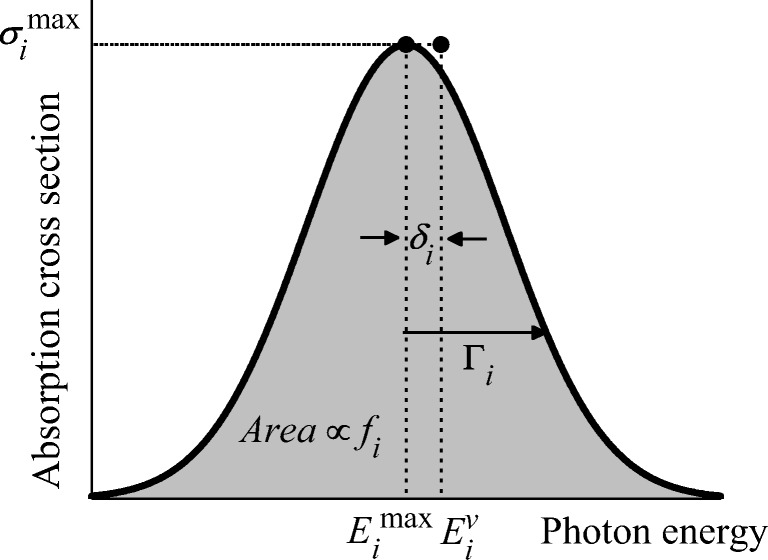
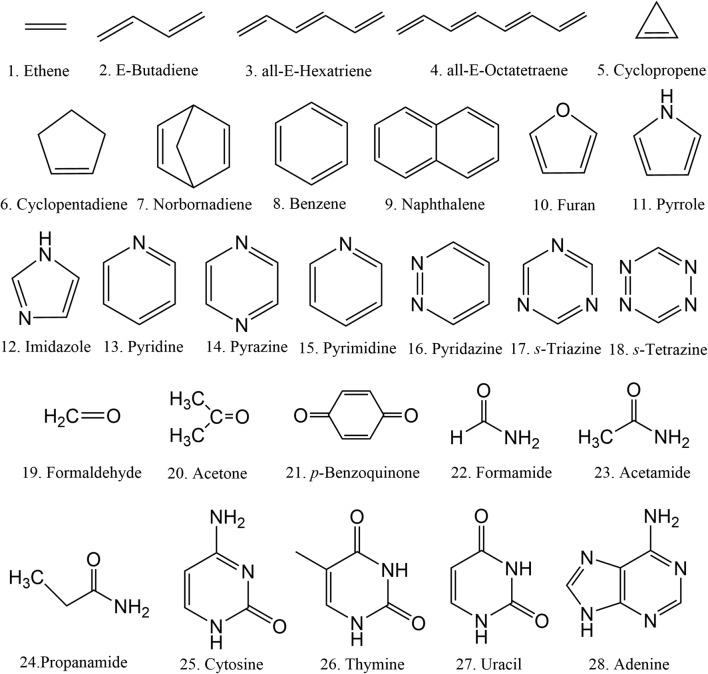
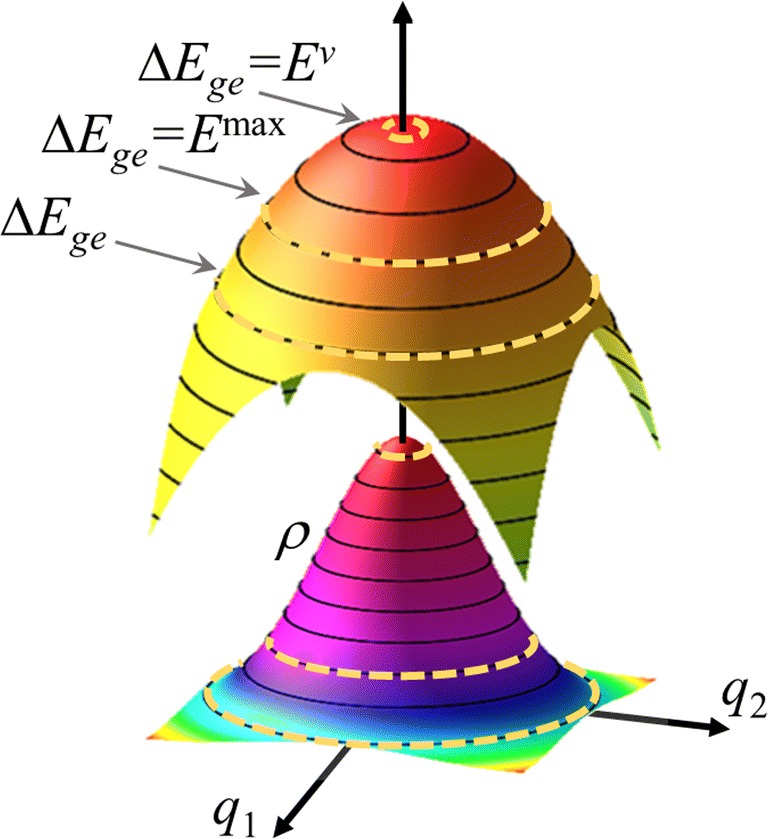
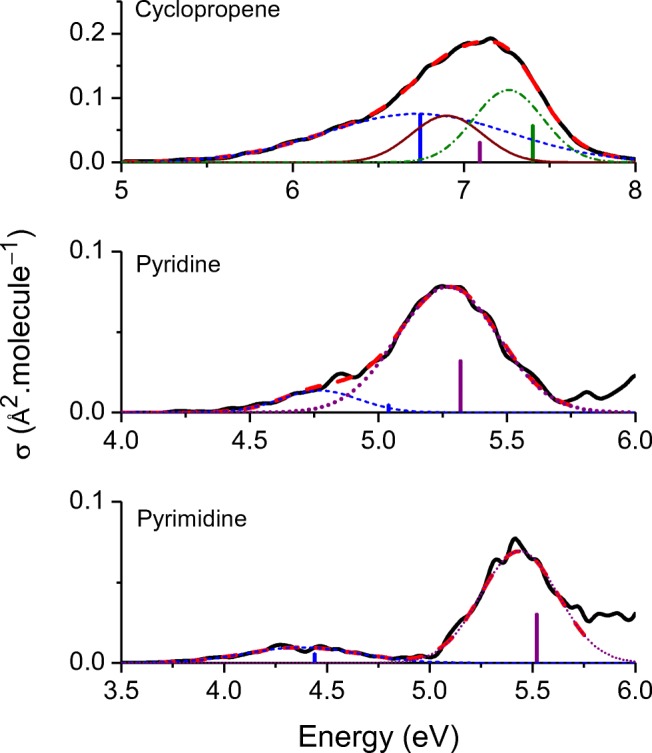
The analysis of the photoabsorption spectra of molecules shows that the band maximum is usually redshifted in comparison to the vertical excitation. We conducted a throughout analysis of this shift based on low-dimensional analytical and numerical model systems, showing that its origin is rooted in the frequency change between the ground and the excited states in multidimensional systems. Moreover, we deliver a benchmark of ab initio results for the shift based on a comparison of vertical excitations and band maxima calculated with the nuclear ensemble approach for the 28 organic molecules in the Mülheim molecular dataset. The mean value of the shift calculated over 60 transitions is 0.11 ± 0.08 eV. The mean value of the band width is 0.32 ± 0.14 eV. Graphical abstract .
分子光吸收光谱分析表明,与垂直激发相比,能带最大值通常会发生红移。我们基于低维分析和数值模型系统对这种位移进行了全面分析,结果表明其起源在于多维系统中基态和激发态之间的频率变化。此外,我们通过比较使用核系综方法计算的穆尔海姆分子数据集中28个有机分子的垂直激发和能带最大值,给出了该位移的从头算结果基准。在60次跃迁上计算得到的位移平均值为0.11±0.08电子伏特。带宽平均值为0.32±0.14电子伏特。图形摘要 。