College of Pharmacy and Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 03760, Korea.
Biomolecules. 2020 Apr 19;10(4):631. doi: 10.3390/biom10040631.
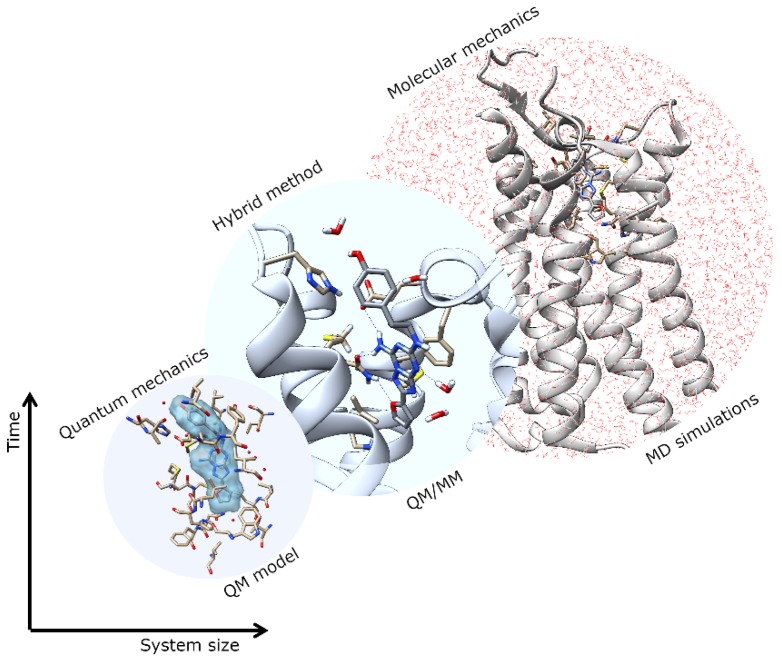
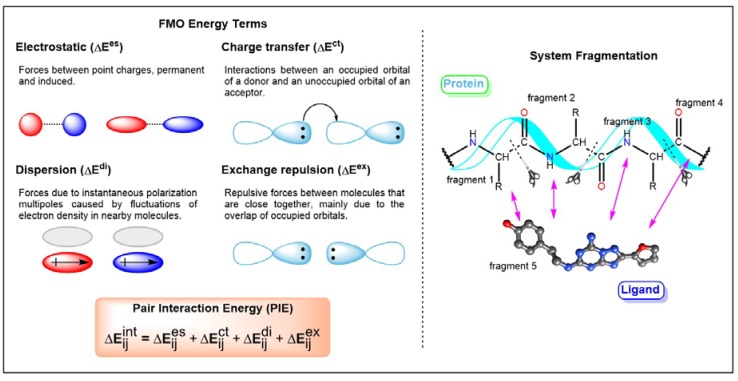
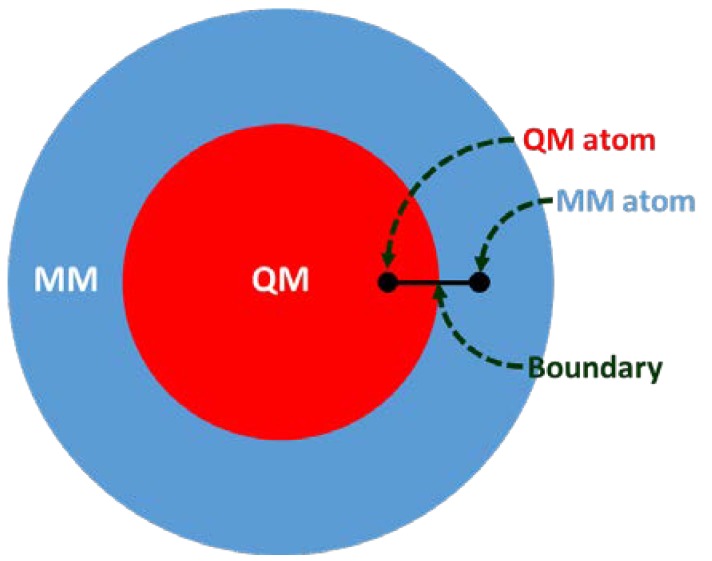
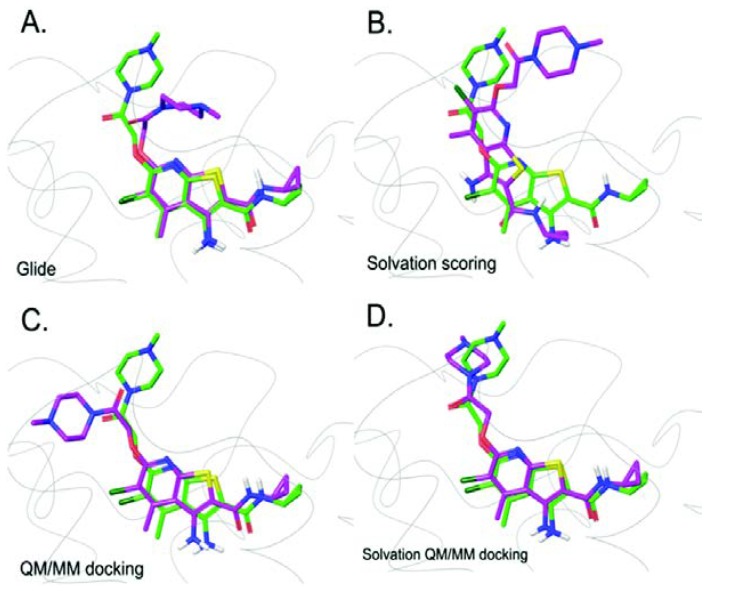
G protein-coupled receptors (GPCRs) are major drug targets due to their ability to facilitate signal transduction across cell membranes, a process that is vital for many physiological functions to occur. The development of computational technology provides modern tools that permit accurate studies of the structures and properties of large chemical systems, such as enzymes and GPCRs, at the molecular level. The advent of multiscale molecular modeling permits the implementation of multiple levels of theories on a system of interest, for instance, assigning chemically relevant regions to high quantum mechanics (QM) level of theory while treating the rest of the system using classical force field (molecular mechanics (MM) potential). Multiscale QM/MM molecular modeling have far-reaching applications in the rational design of GPCR drugs/ligands by affording precise ligand binding configurations through the consideration of conformational plasticity. This enables the identification of key binding site residues that could be targeted to manipulate GPCR function. This review will focus on recent applications of multiscale QM/MM molecular simulations in GPCR studies that could boost the efficiency of future structure-based drug design (SBDD) strategies.
G 蛋白偶联受体(GPCRs)是主要的药物靶点,因为它们能够促进跨细胞膜的信号转导,这是许多生理功能发生所必需的。计算技术的发展为现代工具提供了支持,这些工具允许在分子水平上对酶和 GPCR 等大型化学系统的结构和特性进行准确的研究。多尺度分子建模的出现允许在感兴趣的系统上实施多个层次的理论,例如,将与化学相关的区域分配给高量子力学(QM)理论水平,同时使用经典力场(分子力学(MM)势能)处理系统的其余部分。多尺度 QM/MM 分子建模在 GPCR 药物/配体的合理设计中具有广泛的应用,通过考虑构象可塑性,提供精确的配体结合构象。这能够确定可以靶向以操纵 GPCR 功能的关键结合位点残基。本文将重点介绍多尺度 QM/MM 分子模拟在 GPCR 研究中的最新应用,这将有助于提高基于结构的药物设计(SBDD)策略的效率。