Peng He
School of Information Science and Engineering, Xiamen University, Xiamen, Fujian, China.
PeerJ. 2020 Apr 20;8:e8965. doi: 10.7717/peerj.8965. eCollection 2020.
Conserved nucleic acid sequences play an essential role in transcriptional regulation. The motifs/templates derived from nucleic acid sequence datasets are usually used as biomarkers to predict biochemical properties such as protein binding sites or to identify specific non-coding RNAs. In many cases, template-based nucleic acid sequence classification performs better than some feature extraction methods, such as N-gram and k-spaced pairs classification. The availability of large-scale experimental data provides an unprecedented opportunity to improve motif extraction methods. The process for pattern extraction from large-scale data is crucial for the creation of predictive models.
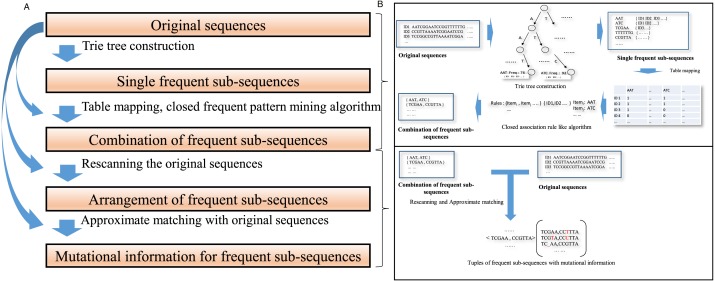
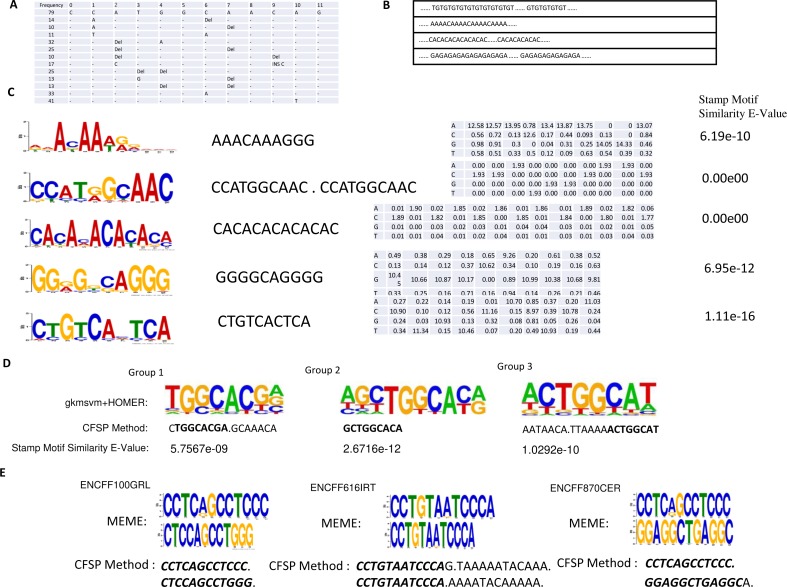
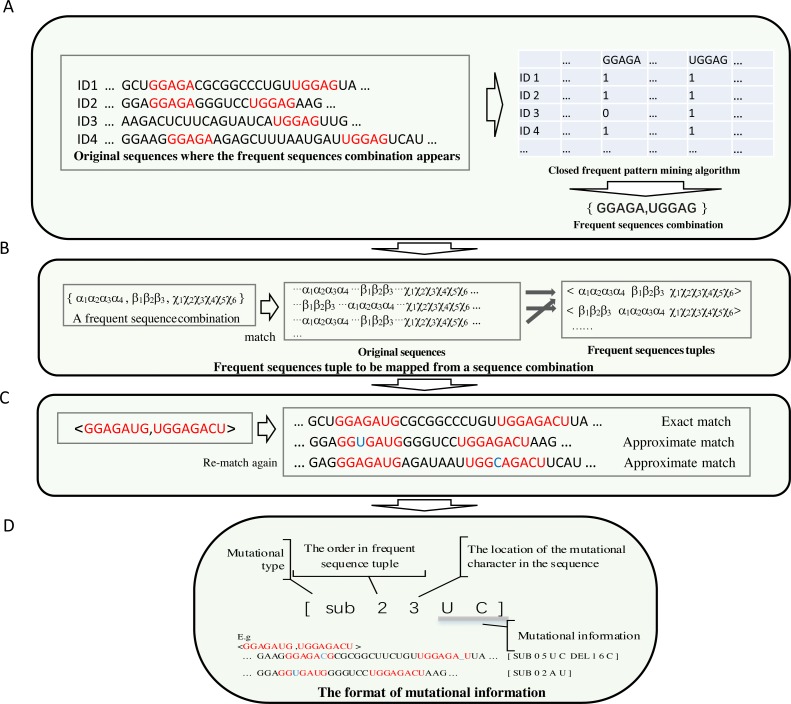
In this article, a Teiresias-like feature extraction algorithm to discover frequent sub-sequences (CFSP) is proposed. Although gaps are allowed in some motif discovery algorithms, the distance and number of gaps are limited. The proposed algorithm can find frequent sequence pairs with a larger gap. The combinations of frequent sub-sequences in given protracted sequences capture the long-distance correlation, which implies a specific molecular biological property. Hence, the proposed algorithm intends to discover the combinations. A set of frequent sub-sequences derived from nucleic acid sequences with order is used as a base frequent sub-sequence array. The mutation information is attached to each sub-sequence array to implement fuzzy matching. Thus, a mutate records a single nucleotide variant or nucleotides insertion/deletion (indel) to encode a slight difference between frequent sequences and a matched subsequence of a sequence under investigation.
The proposed algorithm has been validated with several nucleic acid sequence prediction case studies. These data demonstrate better results than the recently available feature descriptors based methods based on experimental data sets such as miRNA, piRNA, and Sigma 54 promoters. CFSP is implemented in C++ and shell script; the source code and related data are available at https://github.com/HePeng2016/CFSP.
保守核酸序列在转录调控中起着至关重要的作用。从核酸序列数据集中衍生出的基序/模板通常用作生物标志物,以预测诸如蛋白质结合位点等生化特性,或识别特定的非编码RNA。在许多情况下,基于模板的核酸序列分类比某些特征提取方法表现更好,例如N-gram和k间隔对分类。大规模实验数据的可用性为改进基序提取方法提供了前所未有的机会。从大规模数据中提取模式的过程对于创建预测模型至关重要。
本文提出了一种类似提瑞西阿斯的特征提取算法来发现频繁子序列(CFSP)。虽然在一些基序发现算法中允许有间隙,但间隙的距离和数量是有限的。所提出的算法可以找到具有更大间隙的频繁序列对。给定延长序列中频繁子序列的组合捕获了长距离相关性,这意味着特定的分子生物学特性。因此,所提出的算法旨在发现这些组合。一组按顺序从核酸序列衍生的频繁子序列用作基本频繁子序列数组。将突变信息附加到每个子序列数组以实现模糊匹配。因此,一个突变记录一个单核苷酸变体或核苷酸插入/缺失(indel),以编码频繁序列与被研究序列的匹配子序列之间的细微差异。
所提出的算法已通过几个核酸序列预测案例研究得到验证。这些数据表明,与最近基于实验数据集(如miRNA、piRNA和Sigma 54启动子)的基于特征描述符的方法相比,结果更好。CFSP用C++和 shell脚本实现;源代码和相关数据可在https://github.com/HePeng2016/CFSP获得。