Department of Microbiology and Immunology, Indiana University School of Medicine, 2101 E. Coliseum Boulevard, Fort Wayne, IN, 46805, USA.
Department of Anatomy, Cell Biology and Physiology, Indiana University School of Medicine, Fort Wayne, IN, USA.
J Neuroinflammation. 2020 Apr 29;17(1):138. doi: 10.1186/s12974-020-01768-7.
Inflammatory stimuli induce immunoresponsive gene 1 (IRG1) expression that in turn catalyzes the production of itaconate from the tricarboxylic acid cycle. Itaconate has recently emerged as a regulator of immune cell functions, especially in macrophages. Studies show that itaconate is required for the activation of anti-inflammatory transcription factor Nrf2 by LPS in mouse and human macrophages, and LPS-activated IRG1 macrophages that lack endogenous itaconate production exhibit augmented inflammatory responses. Moreover, dimethyl itaconate (DMI), an itaconate derivative, inhibits IL-17-induced IκBς activation in keratinocytes and modulates IL-17-IκBς pathway-mediated skin inflammation in an animal model of psoriasis. Currently, the effect of itaconate on regulating macrophage functions and peripheral inflammatory immune responses is well established. However, its effect on microglia (MG) and CNS inflammatory immune responses remains unexplored. Thus, we investigated whether itaconate possesses an immunomodulatory effect on regulating MG activation and CNS inflammation in animal models of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE).
Chronic C57BL/6 EAE was induced followed by DMI treatment. The effect of DMI on disease severity, blood-brain barrier (BBB) disruption, MG activation, peripheral Th1/Th17 differentiation, and the CNS infiltration of Th1/Th17 cells in EAE was determined. Primary MG was cultured to study the effect of DMI on MG activation. Relapsing-remitting SJL/J EAE was induced to assess the therapeutic effect of DMI.
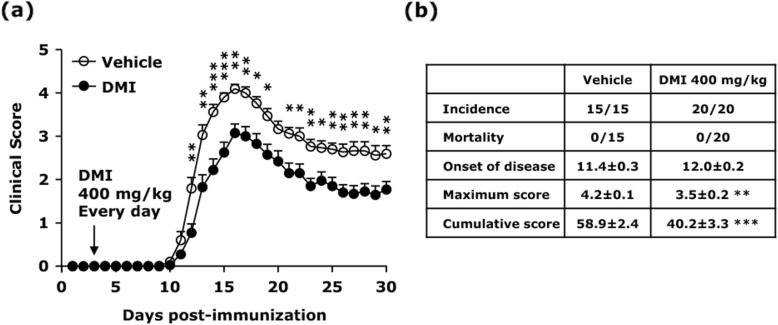
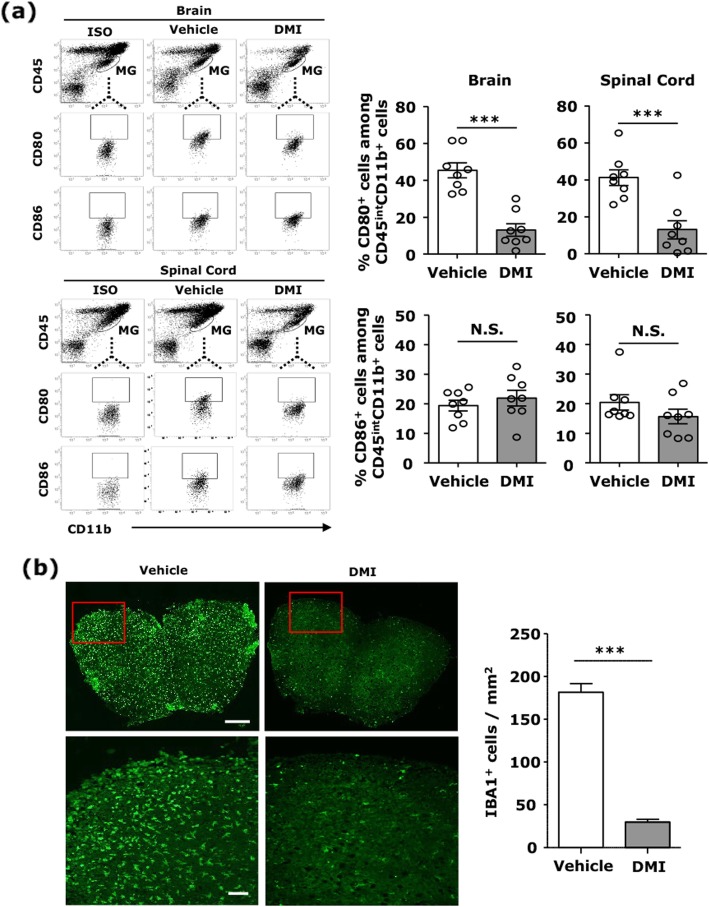
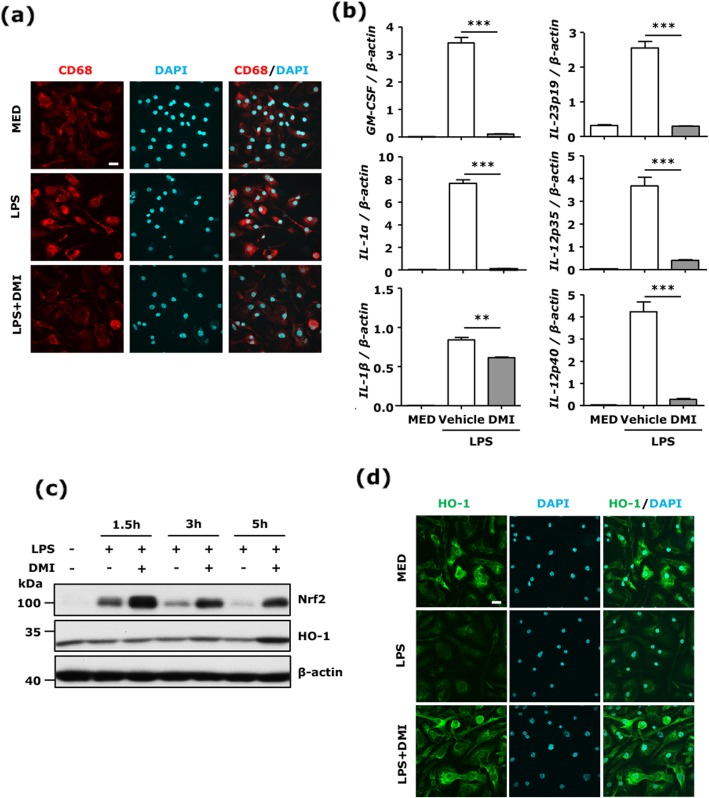
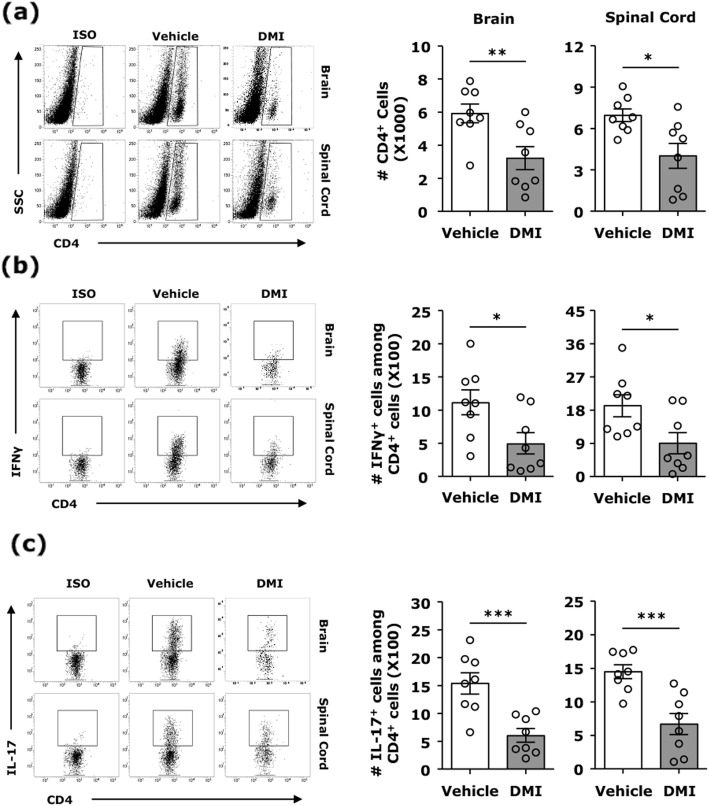
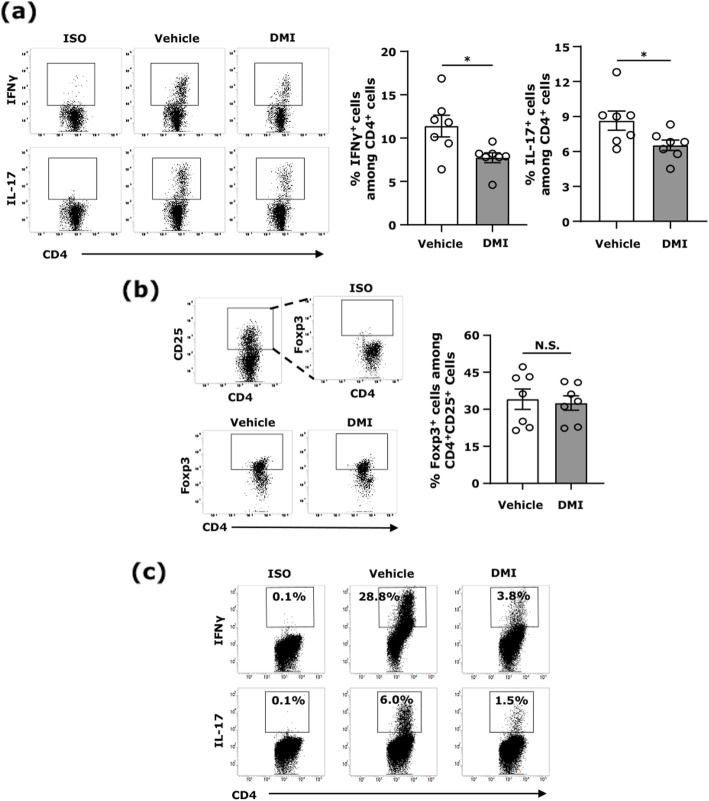
Our results show DMI ameliorated disease severity in the chronic C57BL/6 EAE model. Further analysis of the cellular and molecular mechanisms revealed that DMI mitigated BBB disruption, inhibited MMP3/MMP9 production, suppressed microglia activation, inhibited peripheral Th1/Th17 differentiation, and repressed the CNS infiltration of Th1 and Th17 cells. Strikingly, DMI also exhibited a therapeutic effect on alleviating severity of relapse in the relapsing-remitting SJL/J EAE model.
We demonstrate that DMI suppresses neuroinflammation and ameliorates disease severity in EAE through multiple cellular and molecular mechanisms, suggesting that DMI can be developed as a novel therapeutic agent for the treatment of MS/EAE through its immunomodulatory and anti-inflammatory properties.
炎症刺激诱导免疫反应基因 1(IRG1)表达,进而催化三羧酸循环中衣康酸的生成。衣康酸最近被认为是免疫细胞功能的调节剂,特别是在巨噬细胞中。研究表明,衣康酸是 LPS 在小鼠和人巨噬细胞中激活抗炎转录因子 Nrf2 所必需的,而缺乏内源性衣康酸产生的 LPS 激活的 IRG1 巨噬细胞表现出增强的炎症反应。此外,衣康酸衍生物二甲基衣康酸(DMI)抑制角质形成细胞中白细胞介素-17(IL-17)诱导的 IκBς 激活,并在银屑病动物模型中调节 IL-17-IκBς 通路介导的皮肤炎症。目前,衣康酸对调节巨噬细胞功能和外周炎症免疫反应的作用已得到充分证实。然而,它对小胶质细胞(MG)和中枢神经系统炎症免疫反应的影响仍未得到探索。因此,我们研究了衣康酸是否对多发性硬化症、实验性自身免疫性脑脊髓炎(EAE)的动物模型中的 MG 激活和中枢神经系统炎症具有免疫调节作用。
诱导慢性 C57BL/6 EAE 后进行 DMI 治疗。检测 DMI 对 EAE 疾病严重程度、血脑屏障(BBB)破坏、MG 激活、外周 Th1/Th17 分化以及 EAE 中 Th1/Th17 细胞向中枢神经系统浸润的影响。培养原代 MG 以研究 DMI 对 MG 激活的影响。诱导复发缓解型 SJL/J EAE 以评估 DMI 的治疗效果。
我们的结果表明,DMI 改善了慢性 C57BL/6 EAE 模型的疾病严重程度。进一步的细胞和分子机制分析表明,DMI 减轻了 BBB 破坏,抑制了 MMP3/MMP9 的产生,抑制了小胶质细胞的激活,抑制了外周 Th1/Th17 分化,并抑制了 Th1 和 Th17 细胞向中枢神经系统的浸润。值得注意的是,DMI 还对缓解复发缓解型 SJL/J EAE 模型的严重程度表现出治疗作用。
我们证明,DMI 通过多种细胞和分子机制抑制 EAE 中的神经炎症并改善疾病严重程度,表明 DMI 可以通过其免疫调节和抗炎特性开发为治疗多发性硬化症/实验性自身免疫性脑脊髓炎的新型治疗药物。