Wang Xiaoyuan, Zhang Dongsheng, Zhang Chi, Sun Yueming
The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China.
The Second Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China.
Ann Transl Med. 2020 Mar;8(6):324. doi: 10.21037/atm.2020.02.94.
Abnormal methylation is associated with the survival of colon cancer. This study intended to discover a significant model based on methylation-driven genes (MDGs) and screen relative risk loci to assist with determining the prognoses of colon cancer patients.
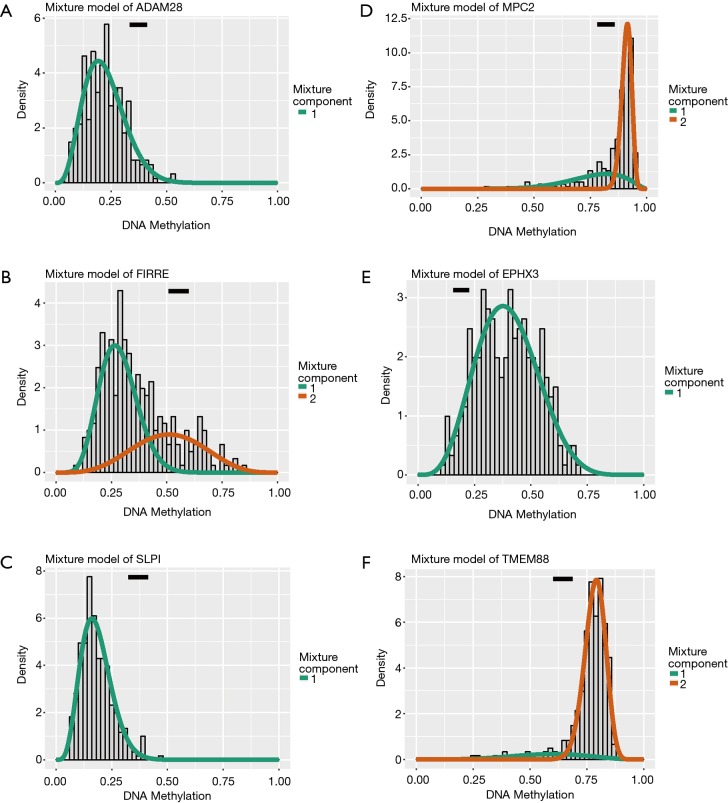
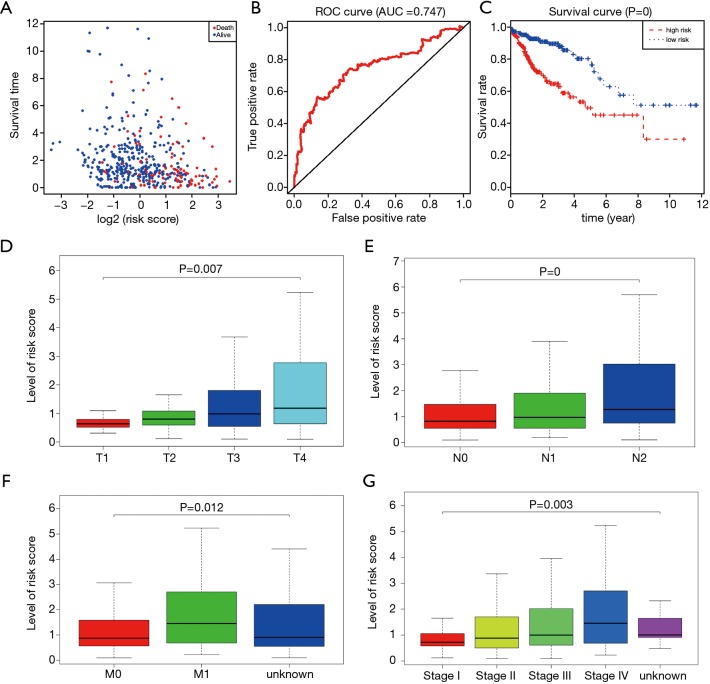
We downloaded transcriptome expression profiles and 450K methylation data from the TCGA database. We then collected the two normalized profiles and utilized the MethylMix package to identify a significant signature showing the aberrantly methylated events highly correlated with expression levels. Also, functional enriched pathway analysis based on the ConsensusPathDB database was conducted to further explore the underlying cancer-related crosstalk among the identified MDGs. To find the significant MDGs for prognosis, we applied a univariate Cox regression model, and the hub signature was identified based on the stepwise regression method. A risk model based on MDGs was constructed from the multivariate Cox analysis, and a receiver operating characteristic (ROC) curve was drawn to assess the predictive value of the MDG signature. Additionally, the Kruskal-Wallis (K-W) test was conducted to compare differential distributions of risk scores across groups of clinical variables. Furthermore, the methylation sites relating to the hub genes were screened out and the prognostic genes were searched using the Cox regression method. Last, we carried out gene set enrichment analysis (GSEA) with the risk score levels serving as the phenotype base on the JAVA platform.
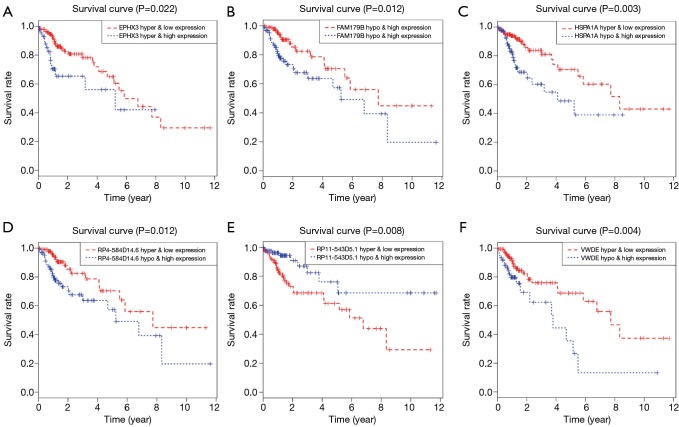
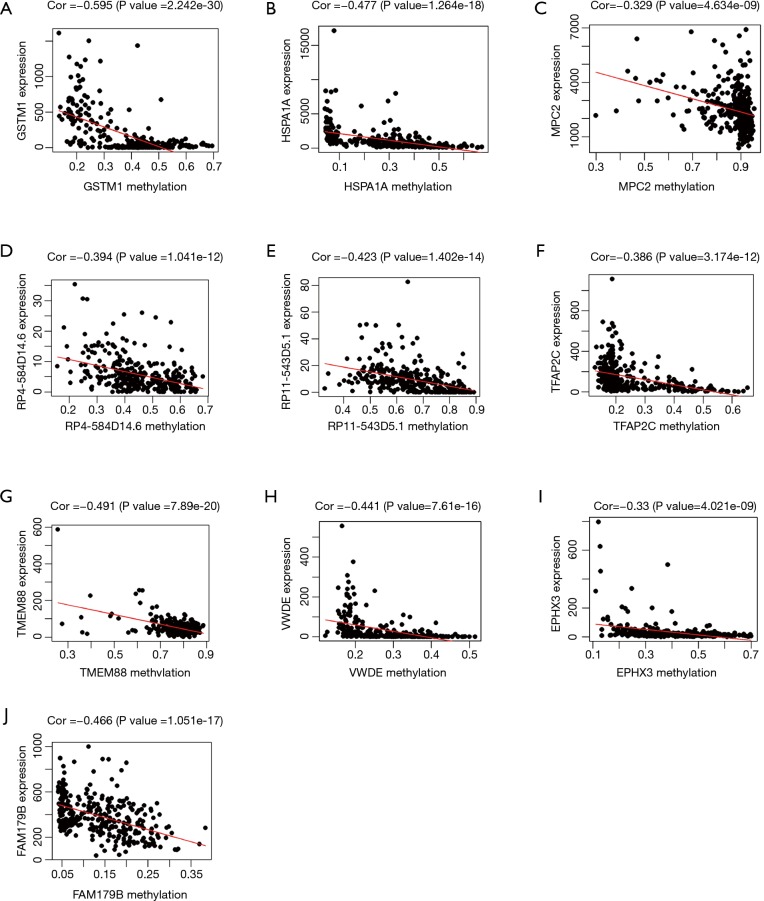
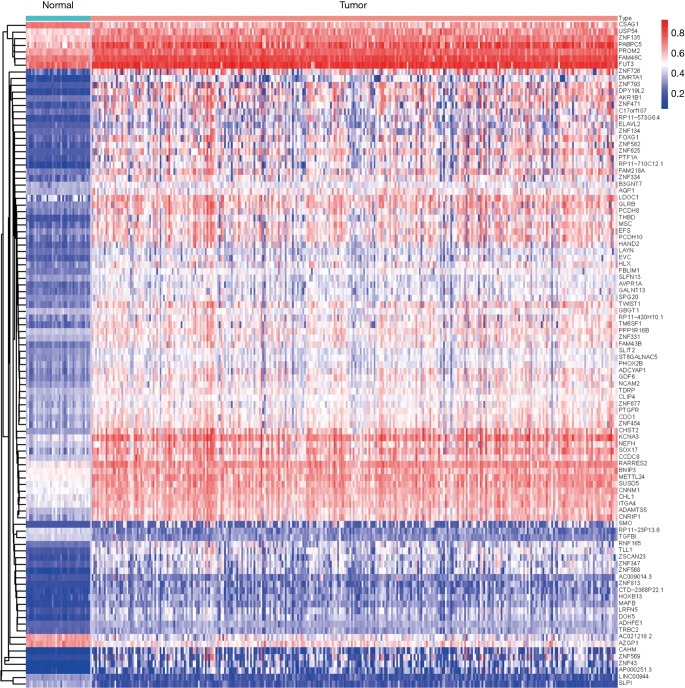
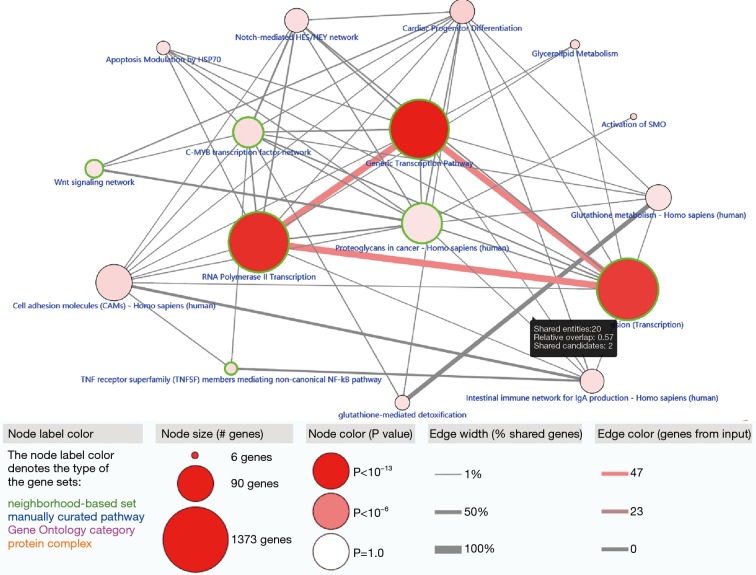
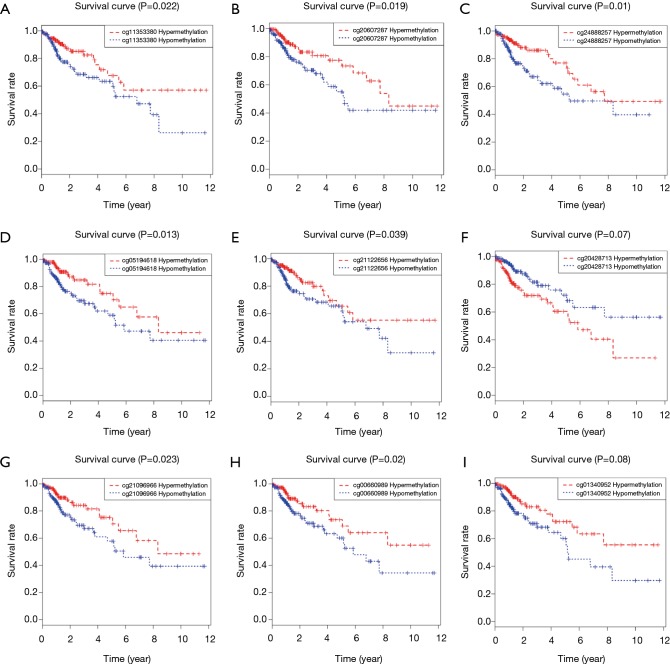
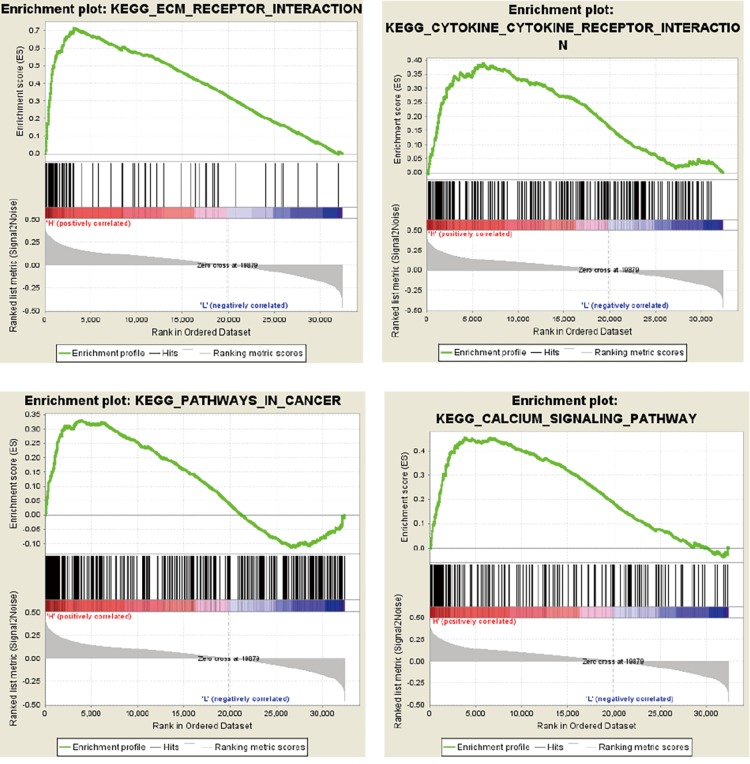
A total of 514 colon cancer samples with transcriptome profiles, including 473 tumor samples and 41 matched normal samples, were downloaded. We also obtained 351 methylation profiles comprising 314 tumor samples and 37 normal samples. The 320 MDGs identified by MethylMix were enriched in the generic transcription pathway, RNA polymerase II transcription, activation of SMO, or glutathione metabolism. Furthermore, a 10-MDGs signature was selected as the hub prognostic marker, and the risk model was constructed from the multivariate Cox regression results. We also discovered multiple specific methylated sites that were highly associated with survival. Finally, the GSEA results suggested that several enriched pathways were associated with the identified risk drivers, including extracellular matrix (ECM) receptor interaction, chemokine receptor interaction, and pathways in cancer, as well as calcium signaling pathways.
We conducted a comprehensive investigation of the molecular mechanisms in colon cancer by discovering the risk methylation-driven signature combined with relative methylated sites and constructing a risk model to predict prognosis.
异常甲基化与结肠癌的生存相关。本研究旨在基于甲基化驱动基因(MDGs)发现一个有意义的模型,并筛选相关风险位点,以协助确定结肠癌患者的预后。
我们从TCGA数据库下载了转录组表达谱和450K甲基化数据。然后收集这两个标准化的谱图,并利用MethylMix软件包来识别一个有意义的特征,该特征显示出与表达水平高度相关的异常甲基化事件。此外,基于ConsensusPathDB数据库进行功能富集通路分析,以进一步探索已识别的MDGs之间潜在的癌症相关相互作用。为了找到用于预后的显著MDGs,我们应用单变量Cox回归模型,并基于逐步回归方法识别出核心特征。基于MDGs构建多变量Cox分析的风险模型,并绘制受试者工作特征(ROC)曲线以评估MDG特征的预测价值。此外,进行Kruskal-Wallis(K-W)检验以比较临床变量组间风险评分的差异分布。此外,筛选出与核心基因相关的甲基化位点,并使用Cox回归方法搜索预后基因。最后,我们在JAVA平台上以风险评分水平作为表型进行基因集富集分析(GSEA)。
共下载了514个具有转录组谱的结肠癌样本,包括473个肿瘤样本和41个匹配的正常样本。我们还获得了351个甲基化谱,包括314个肿瘤样本和37个正常样本。MethylMix识别出的320个MDGs在通用转录途径、RNA聚合酶II转录、SMO激活或谷胱甘肽代谢中富集。此外,选择一个包含10个MDGs的特征作为核心预后标志物,并根据多变量Cox回归结果构建风险模型。我们还发现了多个与生存高度相关的特定甲基化位点。最后,GSEA结果表明,几个富集的通路与已识别的风险驱动因素相关,包括细胞外基质(ECM)受体相互作用、趋化因子受体相互作用、癌症中的通路以及钙信号通路。
我们通过发现风险甲基化驱动的特征并结合相关甲基化位点,对结肠癌的分子机制进行了全面研究,并构建了一个风险模型来预测预后。