Biodiversity Research Center, Academia Sinica, Taipei, Taiwan.
Department of Life Science, National Taiwan Normal University, Taipei, Taiwan.
Sci Rep. 2020 May 5;10(1):7547. doi: 10.1038/s41598-020-64570-8.
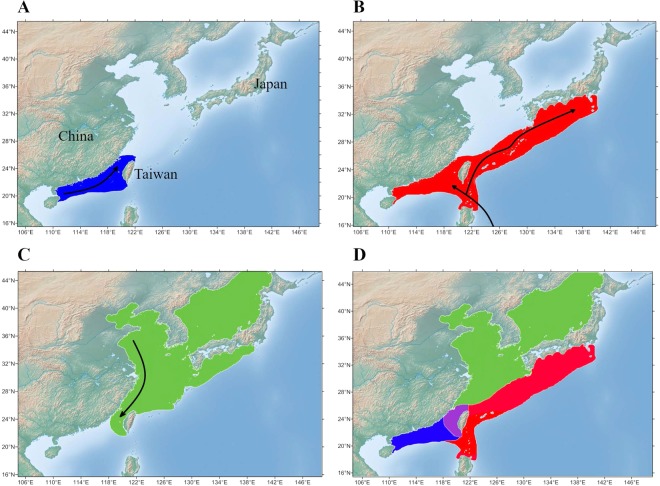
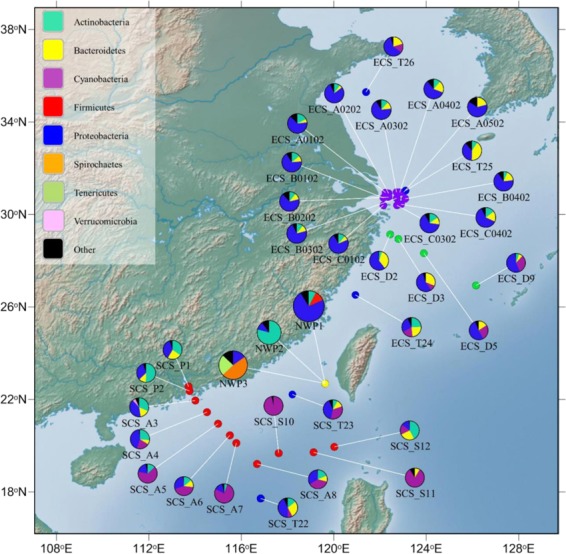
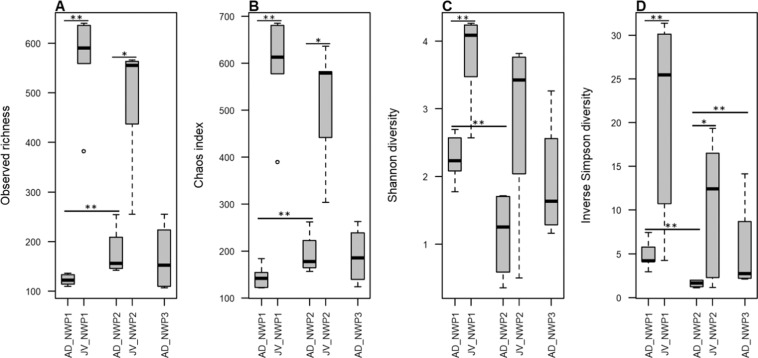
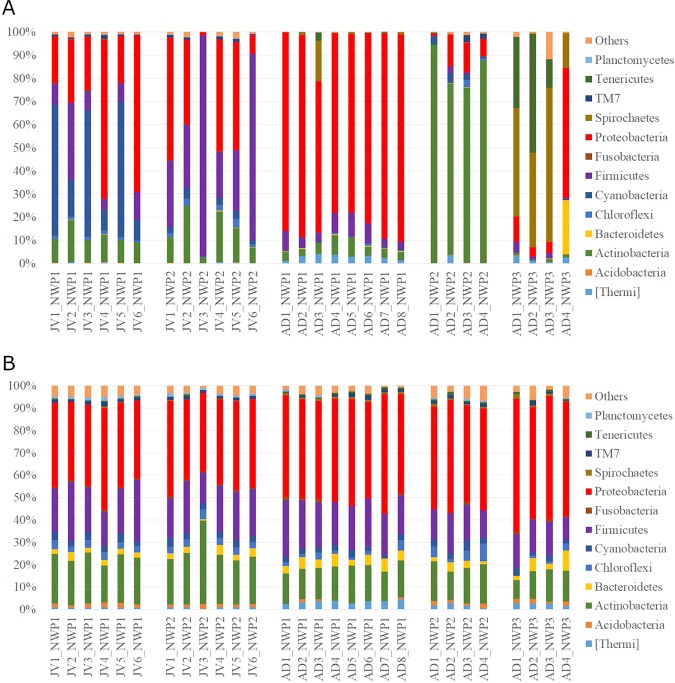
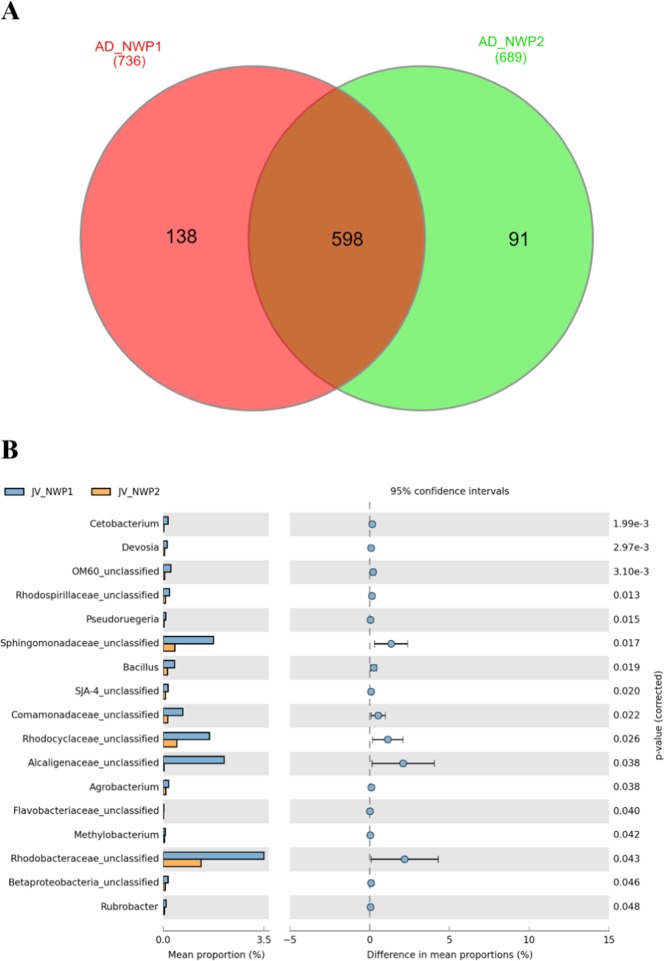
The animal gut microbiota evolves quickly towards a complex community and plays crucial roles in its host's health and development. Factors such as host genetics and environmental changes are regarded as important for controlling the dynamics of animal gut microbiota. Migratory animals are an important group for studying how these factors influence gut microbiota because they experience strong environmental perturbations during migration. The commercially important grey mullet, Mugil cephalus, is a cosmopolitan species complex that display reproductive migration behaviour. There are three cryptic species of M. cephalus fish distributed across the Northwest Pacific, and their spawning sites overlap in the Taiwan Strait. This extraordinary natural occurrence makes the grey mullet an ideal model organism for exploring the nature of wild animal-gut microbiota relationships and interactions. This study investigates the diversity and structure of the gut microbial community in three cryptic M. cephalus species using 16S rRNA amplicon sequencing. Gut microbial compositions from adult and juvenile fish samples were analysed. Our results indicate that gut microbial communities within the grey mullet share a core microbiome dominated by Proteobacteria, Firmicutes and Actinobacteria. However, the structures of gut microbial communities were more distinct between adult mullet groups than they were between juvenile ones. Intriguingly, we found that adult fish that migrate to different geographical tracts harbour gut microbiota similar to historical records of seawater microflora, along their respective migration routes. This observation provides new insights into the interaction between aquatic animal gut microbial communities and the environments along their hosts' migratory routes, and thus warrants future study.
动物肠道微生物群迅速进化为复杂的群落,并在宿主的健康和发育中发挥关键作用。宿主遗传学和环境变化等因素被认为是控制动物肠道微生物群动态的重要因素。迁徙动物是研究这些因素如何影响肠道微生物群的重要群体,因为它们在迁徙过程中经历强烈的环境干扰。商业上重要的灰鲻鱼是一种分布广泛的物种复合体,具有生殖洄游行为。有三种隐蔽的灰鲻鱼分布在西北太平洋,它们的产卵地在台湾海峡重叠。这种非凡的自然发生使灰鲻鱼成为探索野生动物肠道微生物群关系和相互作用本质的理想模式生物。本研究使用 16S rRNA 扩增子测序技术研究了三种隐蔽的灰鲻鱼物种的肠道微生物群落的多样性和结构。分析了成年和幼年鱼类样本的肠道微生物组成。我们的研究结果表明,灰鲻鱼的肠道微生物群落共享一个以变形菌门、厚壁菌门和放线菌门为主的核心微生物组。然而,成年灰鲻鱼群体之间的肠道微生物群落结构比幼年群体之间更为明显。有趣的是,我们发现,沿着各自的洄游路线,迁徙到不同地理区域的成年鱼类,其肠道微生物群类似于历史上记录的海水微生物群。这一观察结果为水生动物肠道微生物群落与宿主洄游路线沿途环境之间的相互作用提供了新的见解,因此值得进一步研究。