Potgieter Lizel, Feurtey Alice, Dutheil Julien Y, Stukenbrock Eva H
Environmental Genomics, Max Planck Institute for Evolutionary Biology, Plön, Germany.
Environmental Genomics, Christian-Albrechts University of Kiel, Kiel, Germany.
Front Microbiol. 2020 Apr 16;11:626. doi: 10.3389/fmicb.2020.00626. eCollection 2020.
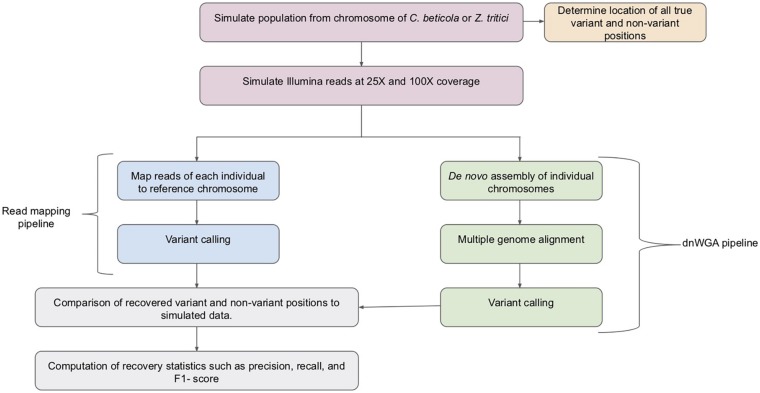
Comparative genome analyses of eukaryotic pathogens including fungi and oomycetes have revealed extensive variability in genome composition and structure. The genomes of individuals from the same population can exhibit different numbers of chromosomes and different organization of chromosomal segments, defining so-called accessory compartments that have been shown to be crucial to pathogenicity in plant-infecting fungi. This high level of structural variation confers a methodological challenge for population genomic analyses. Variant discovery from population sequencing data is typically achieved using established pipelines based on the mapping of short reads to a reference genome. These pipelines have been developed, and extensively used, for eukaryote genomes of both plants and animals, to retrieve single nucleotide polymorphisms and short insertions and deletions. However, they do not permit the inference of large-scale genomic structural variation, as this task typically requires the alignment of complete genome sequences. Here, we compare traditional variant discovery approaches to a pipeline based on genome assembly of short read data followed by whole genome alignment, using simulated data sets with properties mimicking that of fungal pathogen genomes. We show that the latter approach exhibits levels of performance comparable to that of read-mapping based methodologies, when used on sequence data with sufficient coverage. We argue that this approach further allows additional types of genomic diversity to be explored, in particular as long-read third-generation sequencing technologies are becoming increasingly available to generate population genomic data.
对包括真菌和卵菌在内的真核病原体进行的比较基因组分析揭示了基因组组成和结构的广泛变异性。同一群体中个体的基因组可能表现出不同数量的染色体以及染色体片段的不同组织方式,从而定义了所谓的附属区域,这些区域已被证明对植物感染性真菌的致病性至关重要。这种高水平的结构变异给群体基因组分析带来了方法学上的挑战。从群体测序数据中发现变异通常是通过基于将短读段映射到参考基因组的既定流程来实现的。这些流程已经被开发出来并广泛应用于植物和动物的真核生物基因组,以检索单核苷酸多态性以及短插入和缺失。然而,它们不允许推断大规模的基因组结构变异,因为这项任务通常需要完整基因组序列的比对。在这里,我们使用具有模拟真菌病原体基因组特性的模拟数据集,将传统的变异发现方法与基于短读段数据的基因组组装然后进行全基因组比对的流程进行比较。我们表明,当应用于具有足够覆盖度的序列数据时,后一种方法表现出与基于读段映射的方法相当的性能水平。我们认为,这种方法进一步允许探索其他类型的基因组多样性,特别是随着长读段第三代测序技术越来越多地用于生成群体基因组数据。