Lehrstuhl für Allgemeine und Molekulare Botanik, Ruhr-Universität Bochum, Bochum, Germany.
PLoS Genet. 2010 Apr 8;6(4):e1000891. doi: 10.1371/journal.pgen.1000891.
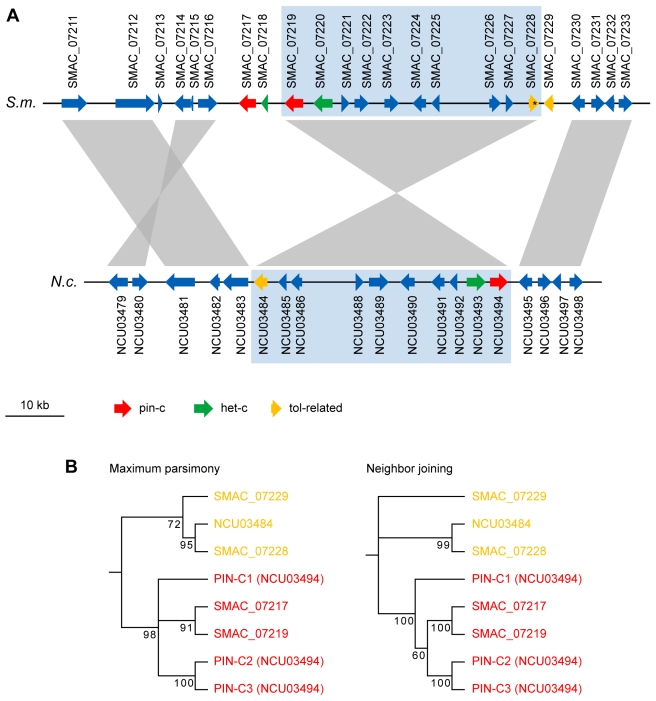
Filamentous fungi are of great importance in ecology, agriculture, medicine, and biotechnology. Thus, it is not surprising that genomes for more than 100 filamentous fungi have been sequenced, most of them by Sanger sequencing. While next-generation sequencing techniques have revolutionized genome resequencing, e.g. for strain comparisons, genetic mapping, or transcriptome and ChIP analyses, de novo assembly of eukaryotic genomes still presents significant hurdles, because of their large size and stretches of repetitive sequences. Filamentous fungi contain few repetitive regions in their 30-90 Mb genomes and thus are suitable candidates to test de novo genome assembly from short sequence reads. Here, we present a high-quality draft sequence of the Sordaria macrospora genome that was obtained by a combination of Illumina/Solexa and Roche/454 sequencing. Paired-end Solexa sequencing of genomic DNA to 85-fold coverage and an additional 10-fold coverage by single-end 454 sequencing resulted in approximately 4 Gb of DNA sequence. Reads were assembled to a 40 Mb draft version (N50 of 117 kb) with the Velvet assembler. Comparative analysis with Neurospora genomes increased the N50 to 498 kb. The S. macrospora genome contains even fewer repeat regions than its closest sequenced relative, Neurospora crassa. Comparison with genomes of other fungi showed that S. macrospora, a model organism for morphogenesis and meiosis, harbors duplications of several genes involved in self/nonself-recognition. Furthermore, S. macrospora contains more polyketide biosynthesis genes than N. crassa. Phylogenetic analyses suggest that some of these genes may have been acquired by horizontal gene transfer from a distantly related ascomycete group. Our study shows that, for typical filamentous fungi, de novo assembly of genomes from short sequence reads alone is feasible, that a mixture of Solexa and 454 sequencing substantially improves the assembly, and that the resulting data can be used for comparative studies to address basic questions of fungal biology.
丝状真菌在生态学、农业、医学和生物技术中具有重要意义。因此,测序了 100 多种丝状真菌的基因组也就不足为奇了,其中大多数是通过桑格测序完成的。虽然下一代测序技术已经彻底改变了基因组重测序,例如用于菌株比较、遗传图谱构建或转录组和 ChIP 分析,但真核生物基因组的从头组装仍然存在重大障碍,这是由于它们的基因组较大且重复序列较多。丝状真菌的 30-90 Mb 基因组中很少有重复区域,因此非常适合通过短序列读取进行从头组装基因组。在这里,我们展示了 Sordaria macrospora 基因组的高质量草图序列,该序列是通过 Illumina/Solexa 和 Roche/454 测序组合获得的。基因组 DNA 的双端 Solexa 测序达到 85 倍覆盖度,另外 10 倍覆盖度是通过单端 454 测序实现的,总共获得了大约 40 Mb 的 DNA 序列。使用 Velvet 组装器将读取组装成一个 40 Mb 的草图版本(N50 为 117 kb)。与 Neurospora 基因组的比较分析将 N50 提高到 498 kb。S. macrospora 基因组的重复区域比其最接近的测序亲缘种 Neurospora crassa 更少。与其他真菌的基因组比较表明,作为形态发生和减数分裂的模式生物,S. macrospora 拥有几个参与自我/非我识别的基因的重复。此外,S. macrospora 比 N. crassa 含有更多的聚酮生物合成基因。系统发育分析表明,其中一些基因可能是通过水平基因转移从远缘的子囊菌获得的。我们的研究表明,对于典型的丝状真菌,仅通过短序列读取进行基因组的从头组装是可行的,Solexa 和 454 测序的组合可以极大地提高组装质量,并且可以使用所得数据进行比较研究,以解决真菌生物学的基本问题。