Lipiński Patryk, Rokicki Dariusz, Bogdańska Anna, Lesiak Justyna, Lefeber Dirk J, Tylki-Szymańska Anna
Department of Pediatrics, Nutrition and Metabolic Diseases The Children's Memorial Health Institute Warsaw Poland.
Department of Biochemistry, Radioimmunology and Experimental Medicine The Children's Memorial Health Institute Warsaw Poland.
JIMD Rep. 2020 Apr 9;53(1):80-82. doi: 10.1002/jmd2.12104. eCollection 2020 May.
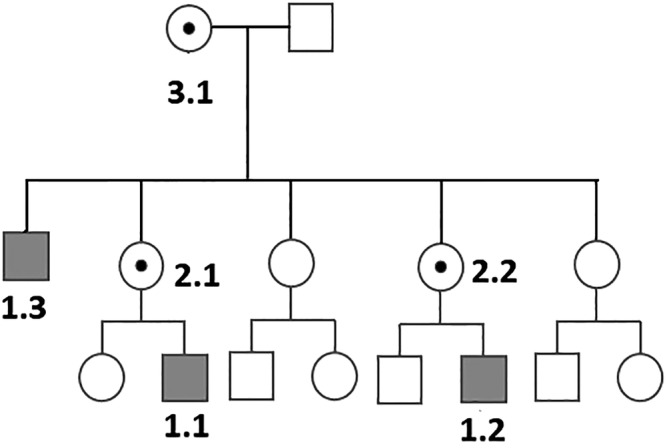
In 2016, 11 male patients were reported with immunodeficiency and hepatic, gastric and (in some) neurological disease due to X-linked ATP6AP1 deficiency (ATP6AP1-CDG). In 2018, three other patients were reported with additional features: connective tissue abnormalities, sensorineural hearing loss, hyperopia, glomerular and tubular dysfunction, exocrine pancreatic insufficiency and altered amino acid and lipid metabolism. We here present a follow-up of three reported siblings showing progression of deafness to total hearing loss, progressive loss of hair up to alopecia, chestnut skin and, at last follow-up, in some of them proteinuria. Three female carriers showed a normal serum transferrin isoelectrofocusing but in two of them there was a persistent proteinuria.
2016年,有报道称11名男性患者因X连锁ATP6AP1缺乏症(ATP6AP1-CDG)出现免疫缺陷以及肝脏、胃部和(部分患者)神经系统疾病。2018年,又有另外3名患者被报道出现其他特征:结缔组织异常、感音神经性听力损失、远视、肾小球和肾小管功能障碍、外分泌性胰腺功能不全以及氨基酸和脂质代谢改变。我们在此展示了对3名已报道的同胞患者的随访情况,结果显示耳聋进展为全聋,毛发逐渐脱落直至秃发,皮肤呈栗色,且在最后一次随访时,部分患者出现蛋白尿。3名女性携带者血清转铁蛋白等电聚焦结果正常,但其中2人存在持续性蛋白尿。