Department of Biochemistry, Radioimmunology and Experimental Medicine, The Children's Memorial Health Institute, Warsaw, Poland.
Department of Pediatrics, Nutrition and Metabolic Diseases, The Children's Memorial Health Institute, Warsaw, Poland.
Orphanet J Rare Dis. 2021 Jan 6;16(1):17. doi: 10.1186/s13023-020-01657-5.
Congenital disorders of glycosylation (CDG) result from defects in the synthesis of glycans and the attachment of glycans to proteins and lipids. Our study aimed to describe the clinical, biochemical, and molecular findings of CDG patients, and to present the long-term follow-up.
A single-center study (1995-2019 years) of patients with congenital disorders of N-glycosylation and combined N- and O-hypoglycosylation was performed.
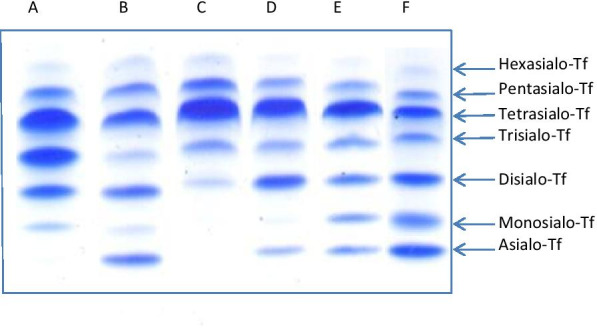
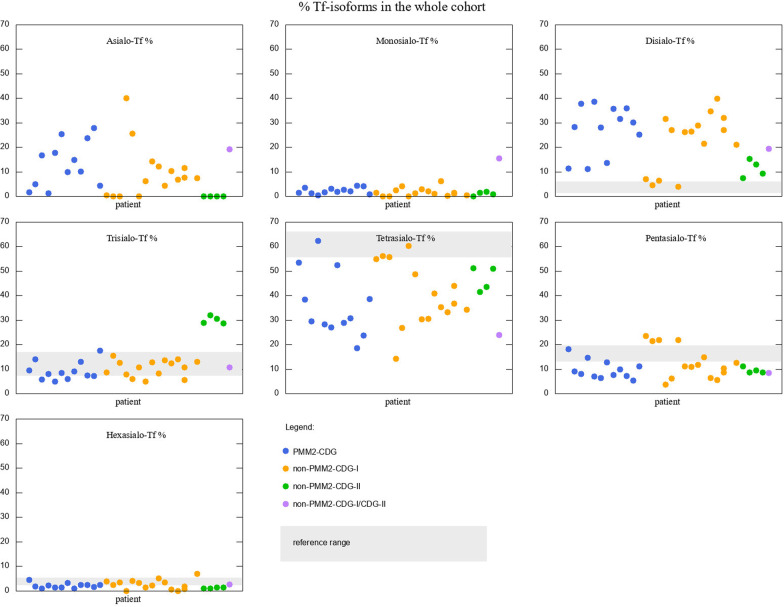
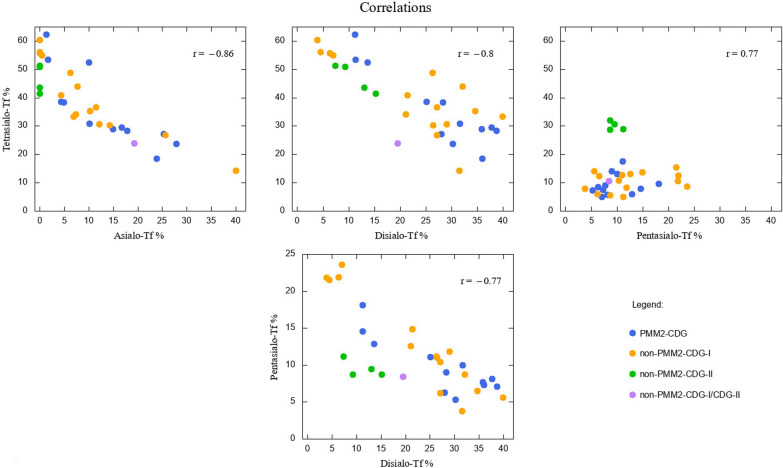
Among 32 patients included into the study, there were 12 PMM2-CDG, 3 ALG13-CDG, 3 ALG1-CDG, 1 ALG3-CDG, 3 MPI-CDG, 1 PGM1-CDG, 4 SRD5A3-CDG, 1 DPAGT1-CDG, 3 ATP6AP1-CDG, 1 ATP6V0A2-CDG. The phenotypic and genotypic spectrum during long-term (in some cases over 20 years) observation was characterised and several measurements of serum Tf isoforms taken. Statistical analysis revealed strong negative correlation between asialo-Tf and tetrasialo-Tf, as well as between disialo-Tf and tetrasialo-Tf. Within CDG type I, no difference in % Tf isoforms was revealed between PMM2-CDG and non-PMM2-CDG patients. However, these two groups differed significantly in such diagnostic features as: cerebellar ataxia, failure to thrive, hypothyroidism, pericardial effusion, cardiomyopathy, inverted nipples, prolonged INR. The effect of treatment with mannose in 2 patients with MPI-CDG was assessed and we found that % of asialo-Tf, monosialo-Tf, and disialo-Tf was significantly lowered, whereas tetrasialo-Tf and pentasialo-Tf rose, coming closer or falling into the reference range.
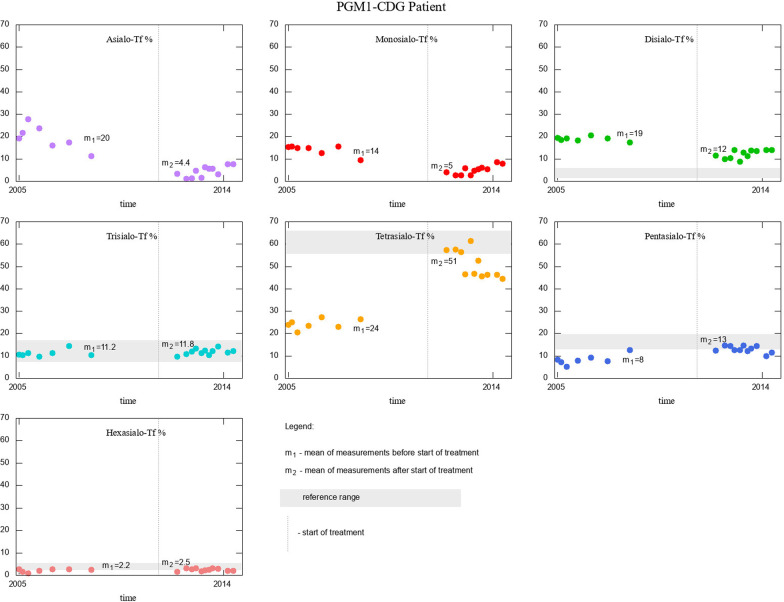
The novel finding was an abnormal Tf IEF pattern in two ALG13-CDG patients and normal in one ALG1-CDG patient. Clinical manifestation of presented CDG patients was similar to that reported in the literature. Mannose supplementation in MPI-CDG patients, as well as galactose supplementation in PGM1-CDG patient, improved patients' clinical picture and Tf isoform profiles.
糖基化先天性疾病(CDG)是由于聚糖的合成缺陷以及聚糖与蛋白质和脂质的连接缺陷引起的。我们的研究旨在描述 CDG 患者的临床、生化和分子发现,并介绍长期随访结果。
对 1995 年至 2019 年期间患有先天性 N-糖基化障碍和 N-及 O-混合糖基化障碍的患者进行了单中心研究。
在纳入研究的 32 名患者中,有 12 名 PMM2-CDG、3 名 ALG13-CDG、3 名 ALG1-CDG、1 名 ALG3-CDG、3 名 MPI-CDG、1 名 PGM1-CDG、4 名 SRD5A3-CDG、1 名 DPAGT1-CDG、3 名 ATP6AP1-CDG、1 名 ATP6V0A2-CDG。在长期(某些情况下超过 20 年)观察期间,对表型和基因型谱进行了特征描述,并对血清转铁蛋白同工型进行了多次测量。统计分析显示,天门冬氨酸转铁蛋白的无唾液酸同工型与四唾液酸同工型之间以及二唾液酸同工型与四唾液酸同工型之间存在强烈的负相关。在 CDG Ⅰ型中,PMM2-CDG 与非 PMM2-CDG 患者之间的转铁蛋白同工型%无差异。然而,这两组在以下诊断特征上存在显著差异:小脑共济失调、生长不良、甲状腺功能减退、心包积液、心肌病、内陷乳头、INR 延长。评估了 2 名 MPI-CDG 患者甘露糖治疗的效果,我们发现无唾液酸转铁蛋白、单唾液酸转铁蛋白和二唾液酸转铁蛋白的%显著降低,而四唾液酸转铁蛋白和五唾液酸转铁蛋白升高,接近或落入参考范围。
新发现是两名 ALG13-CDG 患者的异常转铁蛋白等电聚焦图谱和一名 ALG1-CDG 患者的正常图谱。本研究中 CDG 患者的临床表现与文献报道相似。MPI-CDG 患者补充甘露糖以及 PGM1-CDG 患者补充半乳糖可改善患者的临床症状和转铁蛋白同工型谱。