School of Computer Science and Engineering, Pusan National University, Busan, 46241, South Korea.
Department of Genetics, Stanford University, Stanford, 94305, USA.
Sci Rep. 2020 May 13;10(1):7933. doi: 10.1038/s41598-020-64655-4.
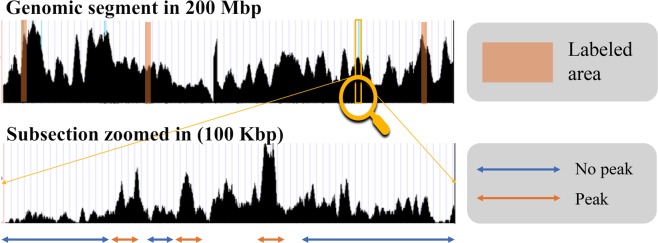
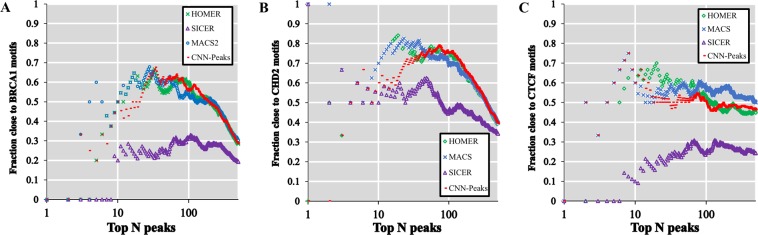
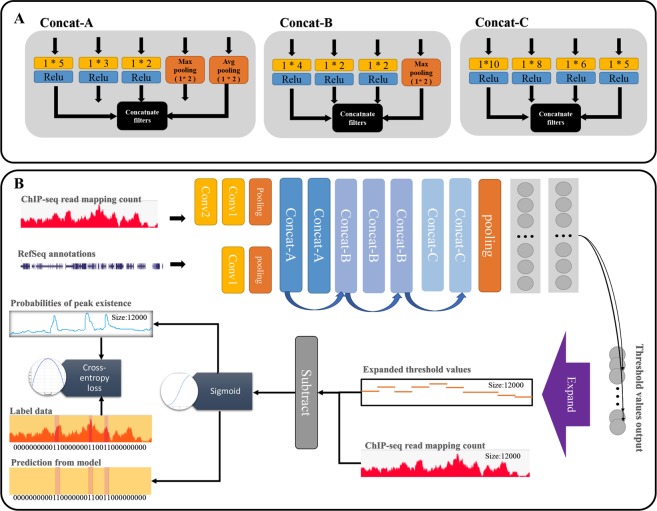
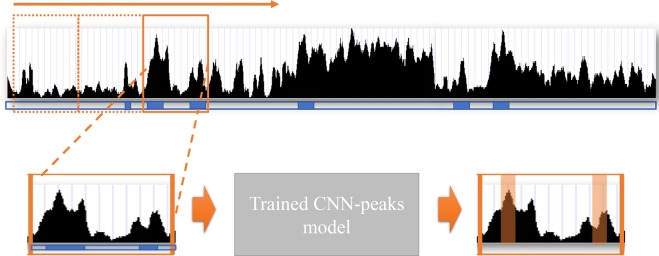
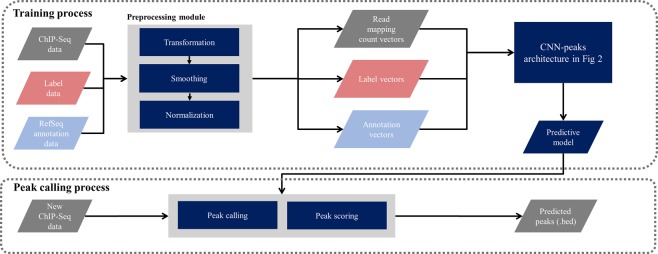
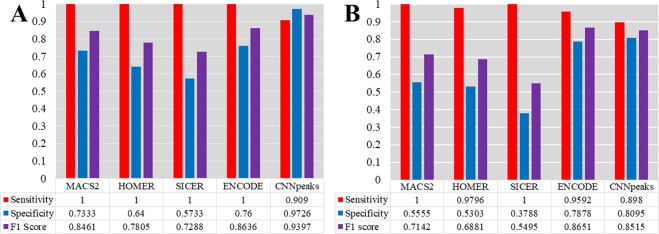
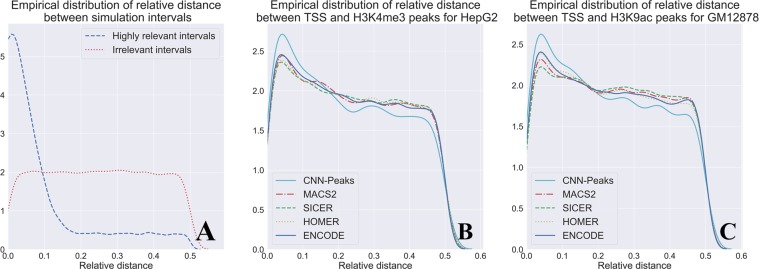
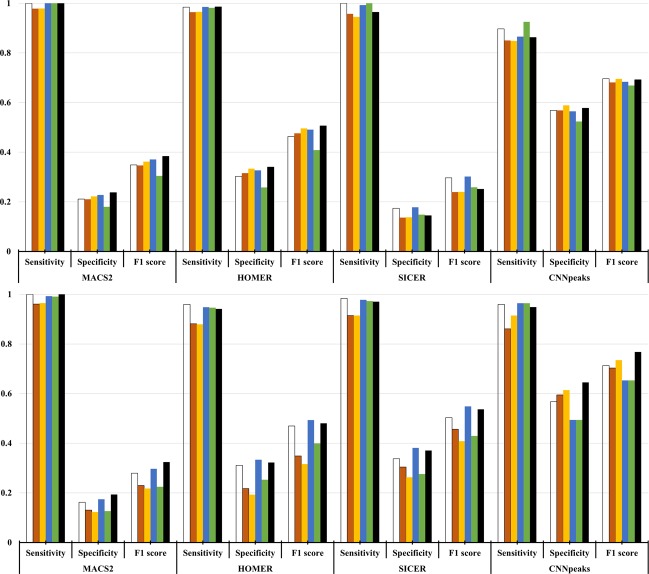
ChIP-seq is one of the core experimental resources available to understand genome-wide epigenetic interactions and identify the functional elements associated with diseases. The analysis of ChIP-seq data is important but poses a difficult computational challenge, due to the presence of irregular noise and bias on various levels. Although many peak-calling methods have been developed, the current computational tools still require, in some cases, human manual inspection using data visualization. However, the huge volumes of ChIP-seq data make it almost impossible for human researchers to manually uncover all the peaks. Recently developed convolutional neural networks (CNN), which are capable of achieving human-like classification accuracy, can be applied to this challenging problem. In this study, we design a novel supervised learning approach for identifying ChIP-seq peaks using CNNs, and integrate it into a software pipeline called CNN-Peaks. We use data labeled by human researchers who annotate the presence or absence of peaks in some genomic segments, as training data for our model. The trained model is then applied to predict peaks in previously unseen genomic segments from multiple ChIP-seq datasets including benchmark datasets commonly used for validation of peak calling methods. We observe a performance superior to that of previous methods.
ChIP-seq 是一种用于了解全基因组表观遗传相互作用并识别与疾病相关的功能元件的核心实验资源。ChIP-seq 数据的分析很重要,但由于存在各种级别的不规则噪声和偏差,因此具有一定的计算挑战性。尽管已经开发了许多峰调用方法,但当前的计算工具在某些情况下仍然需要使用数据可视化进行人工手动检查。然而,庞大的 ChIP-seq 数据量使得人类研究人员几乎不可能手动发现所有的峰。最近开发的卷积神经网络(CNN),能够达到类似人类的分类准确性,可以应用于这个具有挑战性的问题。在这项研究中,我们设计了一种使用 CNN 识别 ChIP-seq 峰的新型监督学习方法,并将其集成到一个名为 CNN-Peaks 的软件管道中。我们使用人类研究人员标记的、在某些基因组片段中存在或不存在峰的数据作为训练数据。然后,将训练好的模型应用于从多个 ChIP-seq 数据集(包括常用的峰调用方法验证基准数据集)中预测以前未见过的基因组片段中的峰。我们观察到的性能优于以前的方法。