SynBioC Research Group, Department of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, B-9000, Ghent, Belgium.
Division of Clinical Pharmacology, Department of Medicine, Medical School, University of Cape Town, K45, OMB, Groote Schuur Hospital, Observatory, 7925, South Africa; Wellcome Centre for Infectious Diseases Research in Africa, Institute of Infectious Disease and Molecular Medicine, South Africa.
Eur J Med Chem. 2020 Jul 15;198:112330. doi: 10.1016/j.ejmech.2020.112330. Epub 2020 Apr 23.
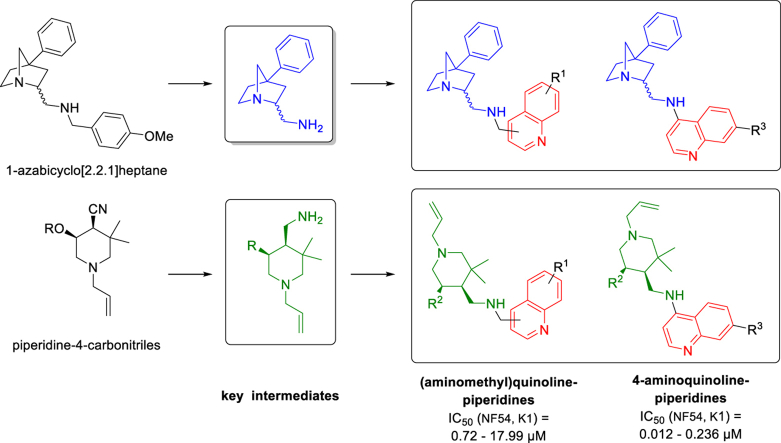
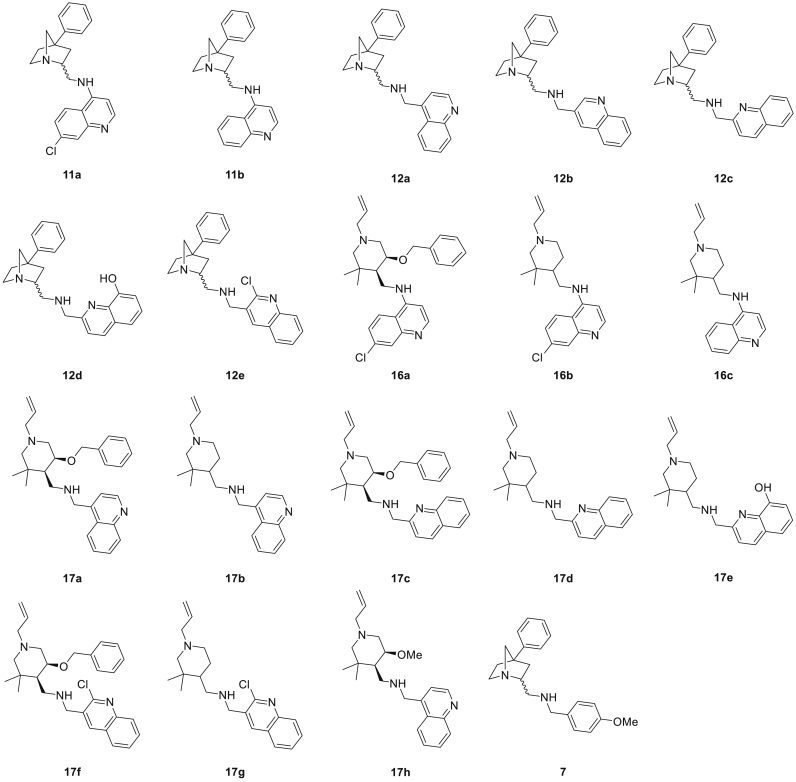
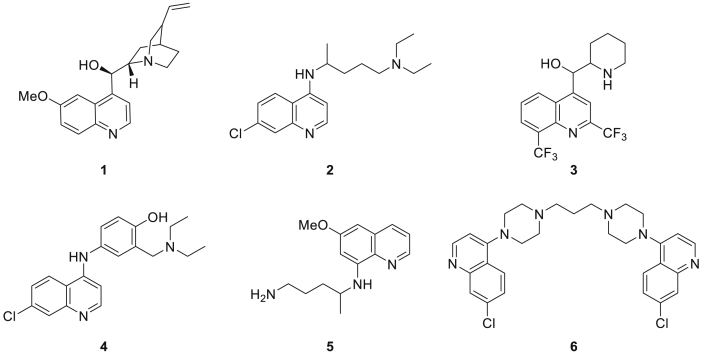
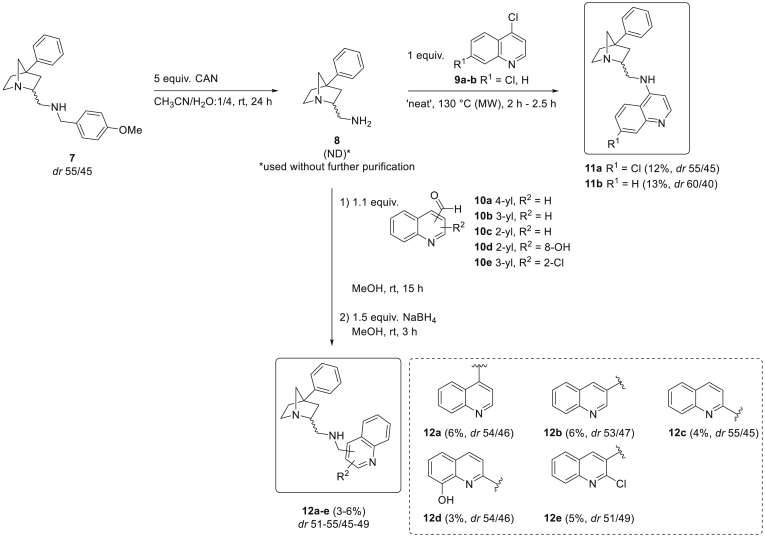
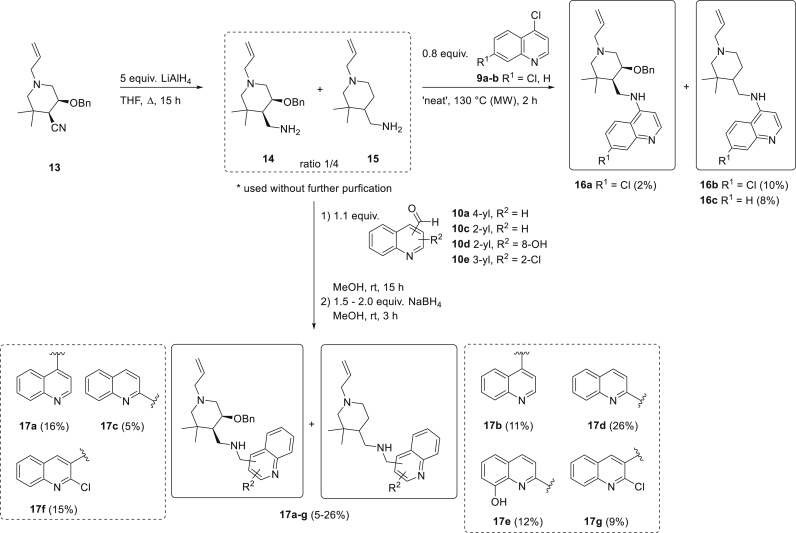
The parasitic disease malaria places almost half of the world's population at risk of infection and is responsible for more than 400,000 deaths each year. The first-line treatment, artemisinin combination therapies (ACT) regimen, is under threat due to emerging resistance of Plasmodium falciparum strains in e.g. the Mekong delta. Therefore, the development of new antimalarial agents is crucial in order to circumvent the growing resistance. Chloroquine, the long-established antimalarial drug, still serves as model compound for the design of new quinoline analogues, resulting in numerous new active derivatives against chloroquine-resistant P. falciparum strains over the past twenty years. In this work, a set of functionalized quinoline analogues, decorated with a modified piperidine-containing side chain, was synthesized. Both amino- and (aminomethyl)quinolines were prepared, resulting in a total of 18 novel quinoline-piperidine conjugates representing four different chemical series. Evaluation of their in vitro antiplasmodium activity against a CQ-sensitive (NF54) and a CQ-resistant (K1) strain of P. falciparum unveiled highly potent activities in the nanomolar range against both strains for five 4-aminoquinoline derivatives. Moreover, no cytotoxicity was observed for all active compounds at the maximum concentration tested. These five new aminoquinoline hit structures are therefore of considerable value for antimalarial research and have the potency to be transformed into novel antimalarial agents upon further hit-to-lead optimization studies.
寄生虫病疟疾使世界近一半人口面临感染风险,每年导致超过 40 万人死亡。一线治疗方法青蒿素联合疗法(ACT)因恶性疟原虫菌株在湄公河三角洲等地出现抗药性而受到威胁。因此,开发新的抗疟药物对于规避日益严重的抗药性至关重要。氯喹是一种历史悠久的抗疟药物,仍然是设计新型喹啉类似物的模型化合物,在过去二十年中,针对氯喹耐药的恶性疟原虫菌株,设计出了许多新的喹啉类似物。在这项工作中,合成了一组带有修饰哌啶侧链的功能化喹啉类似物。制备了氨基和(氨甲基)喹啉,总共合成了 18 种新型的喹啉-哌啶缀合物,代表四个不同的化学系列。评估它们对 CQ 敏感(NF54)和 CQ 耐药(K1)株恶性疟原虫的体外抗疟活性,发现 5 种 4-氨基喹啉衍生物对两种菌株均具有纳摩尔级的高效活性。此外,在测试的最大浓度下,所有活性化合物均未观察到细胞毒性。因此,这 5 种新的氨基喹啉结构对于抗疟研究具有重要价值,并且有潜力在进一步的基于命中的先导优化研究中转化为新型抗疟药物。