Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas, Texas, United States of America.
Departments of Biophysics and Biochemistry, University of Texas Southwestern Medical Center, Dallas, Texas, United States of America.
PLoS Comput Biol. 2020 May 15;16(5):e1007775. doi: 10.1371/journal.pcbi.1007775. eCollection 2020 May.
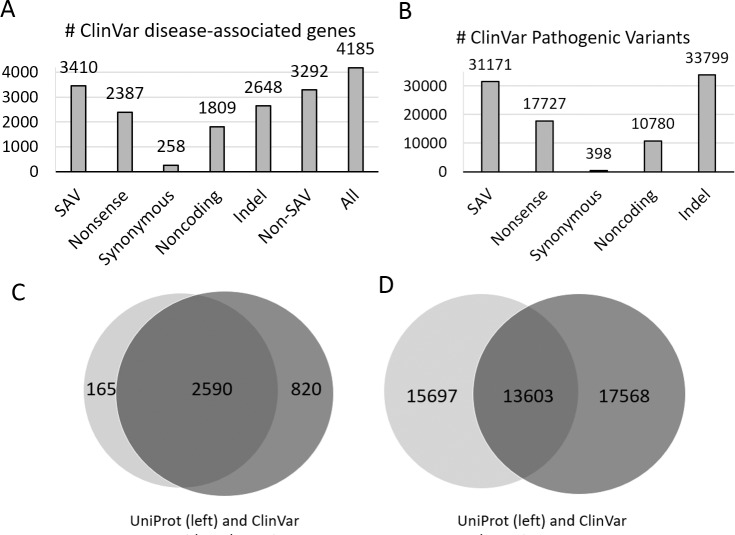
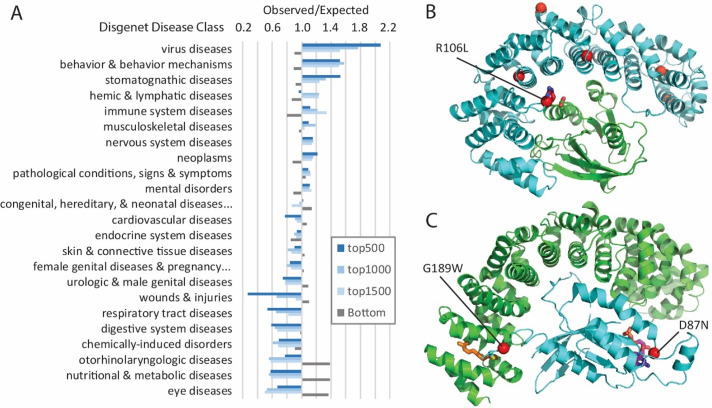
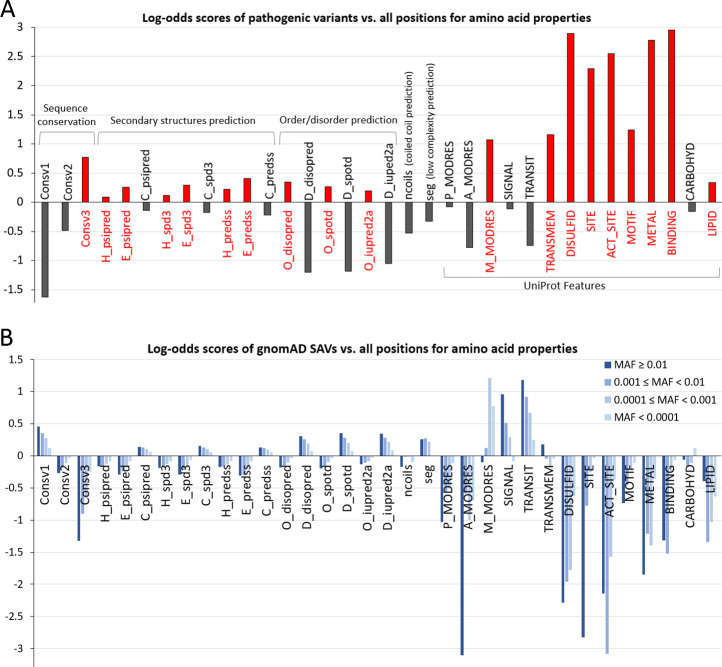
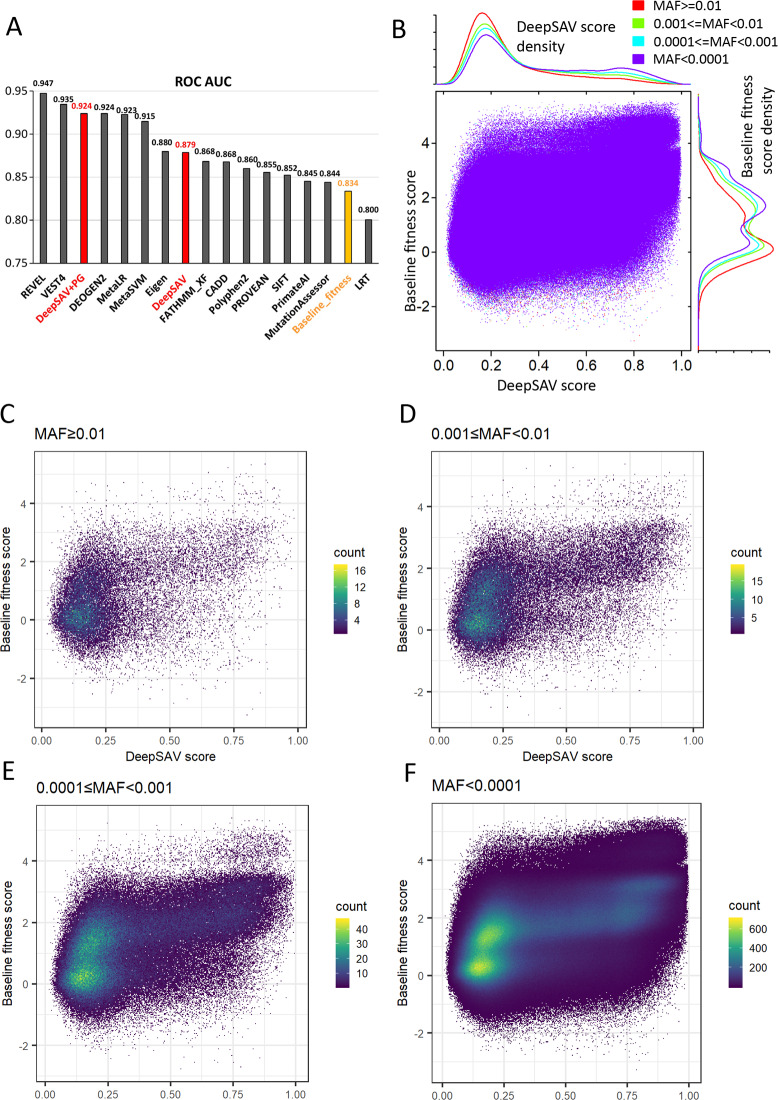
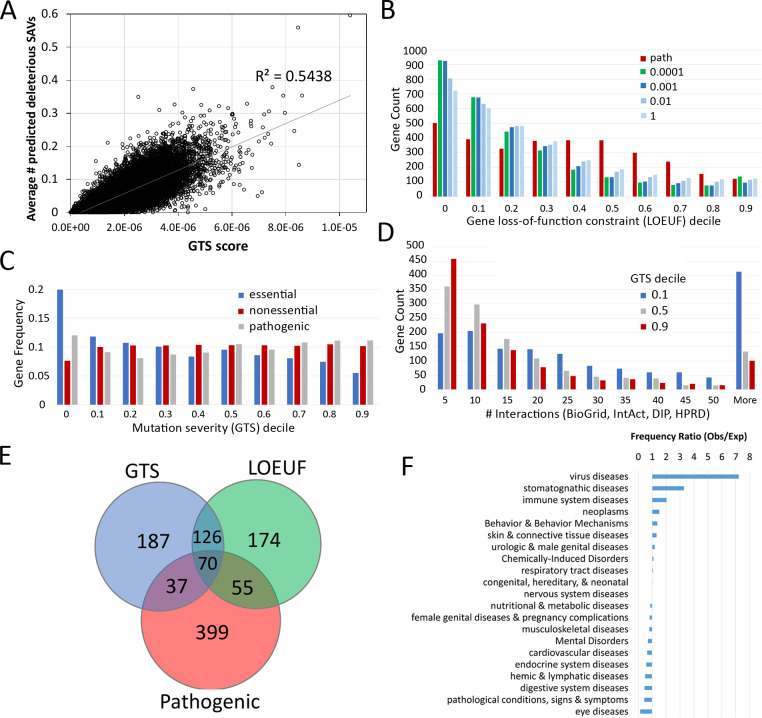
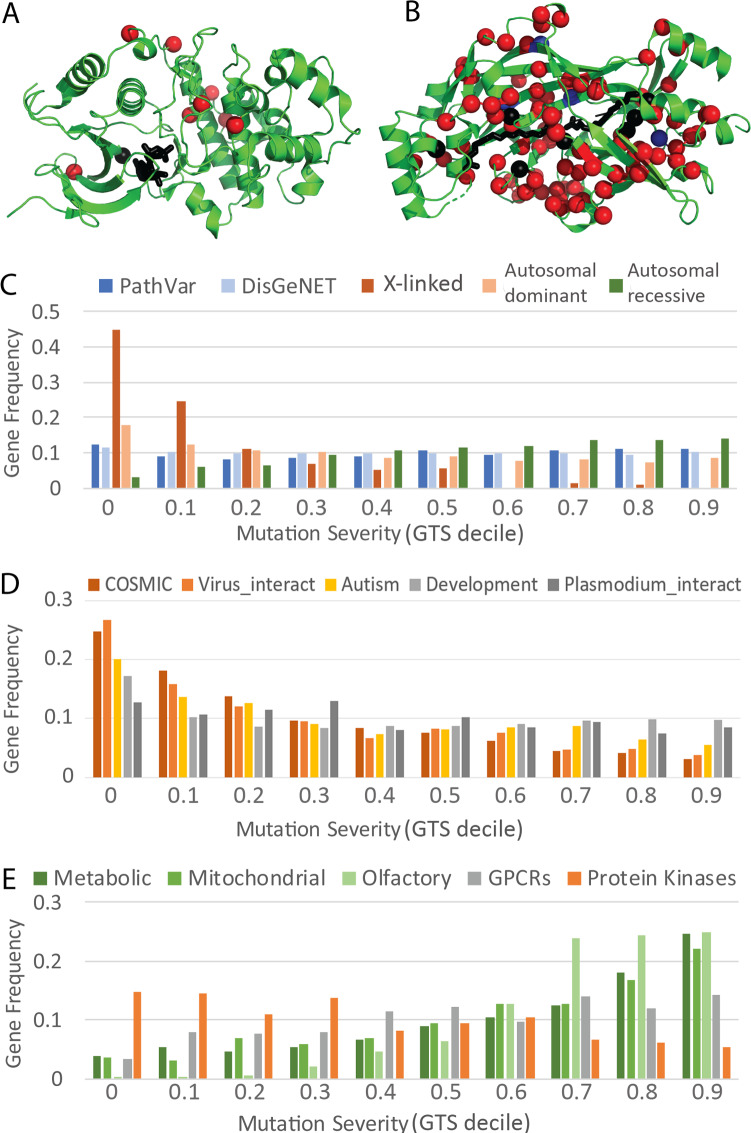
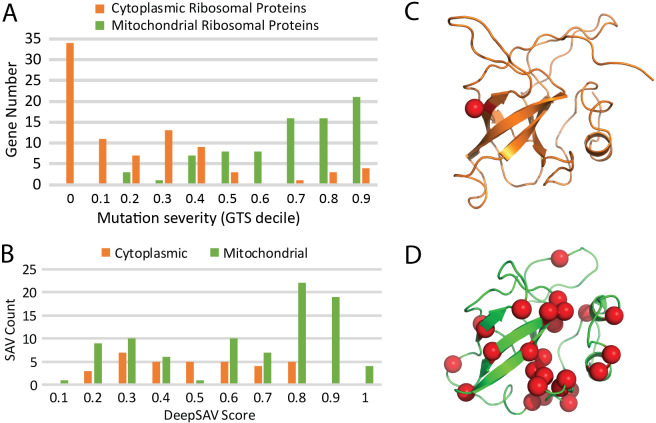
The human genome harbors a variety of genetic variations. Single-nucleotide changes that alter amino acids in protein-coding regions are one of the major causes of human phenotypic variation and diseases. These single-amino acid variations (SAVs) are routinely found in whole genome and exome sequencing. Evaluating the functional impact of such genomic alterations is crucial for diagnosis of genetic disorders. We developed DeepSAV, a deep-learning convolutional neural network to differentiate disease-causing and benign SAVs based on a variety of protein sequence, structural and functional properties. Our method outperforms most stand-alone programs, and the version incorporating population and gene-level information (DeepSAV+PG) has similar predictive power as some of the best available. We transformed DeepSAV scores of rare SAVs in the human population into a quantity termed "mutation severity measure" for each human protein-coding gene. It reflects a gene's tolerance to deleterious missense mutations and serves as a useful tool to study gene-disease associations. Genes implicated in cancer, autism, and viral interaction are found by this measure as intolerant to mutations, while genes associated with a number of other diseases are scored as tolerant. Among known disease-associated genes, those that are mutation-intolerant are likely to function in development and signal transduction pathways, while those that are mutation-tolerant tend to encode metabolic and mitochondrial proteins.
人类基因组中存在多种遗传变异。改变蛋白质编码区域中氨基酸的单核苷酸变化是人类表型变异和疾病的主要原因之一。这些单氨基酸变异(SAVs)在全基因组和外显子组测序中经常被发现。评估这些基因组改变的功能影响对于遗传疾病的诊断至关重要。我们开发了 DeepSAV,这是一种基于多种蛋白质序列、结构和功能特性的深度学习卷积神经网络,用于区分致病和良性 SAVs。我们的方法优于大多数独立程序,而整合了人群和基因水平信息的版本(DeepSAV+PG)与一些最可用的程序具有相似的预测能力。我们将人群中罕见 SAV 的 DeepSAV 分数转化为每个人类蛋白质编码基因的“突变严重程度度量”。它反映了一个基因对有害错义突变的容忍度,是研究基因-疾病关联的有用工具。通过这种衡量标准,发现了与癌症、自闭症和病毒相互作用相关的基因对突变不敏感,而与许多其他疾病相关的基因则被评为耐受。在已知的与疾病相关的基因中,那些突变不敏感的基因可能在发育和信号转导途径中起作用,而那些突变耐受的基因则倾向于编码代谢和线粒体蛋白。