Department CIBIO, University of Trento, Trento, Italy.
Department of Pediatrics, University of California San Diego, La Jolla, CA, USA.
Nat Commun. 2020 May 19;11(1):2500. doi: 10.1038/s41467-020-16366-7.
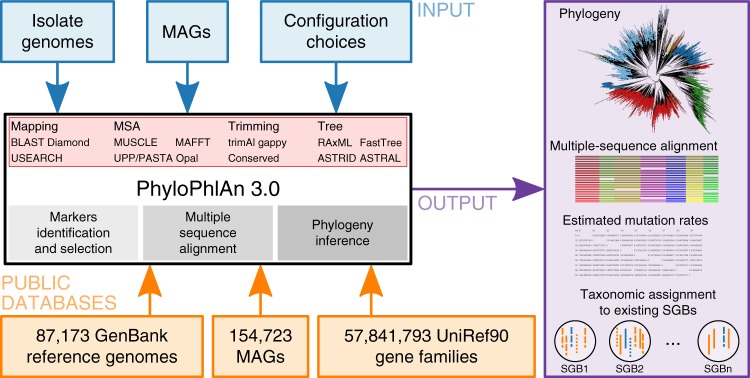
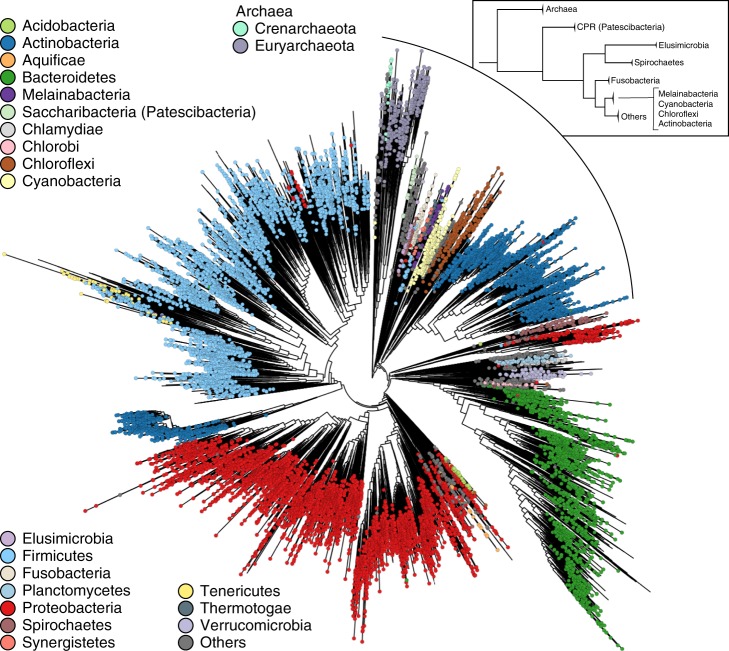
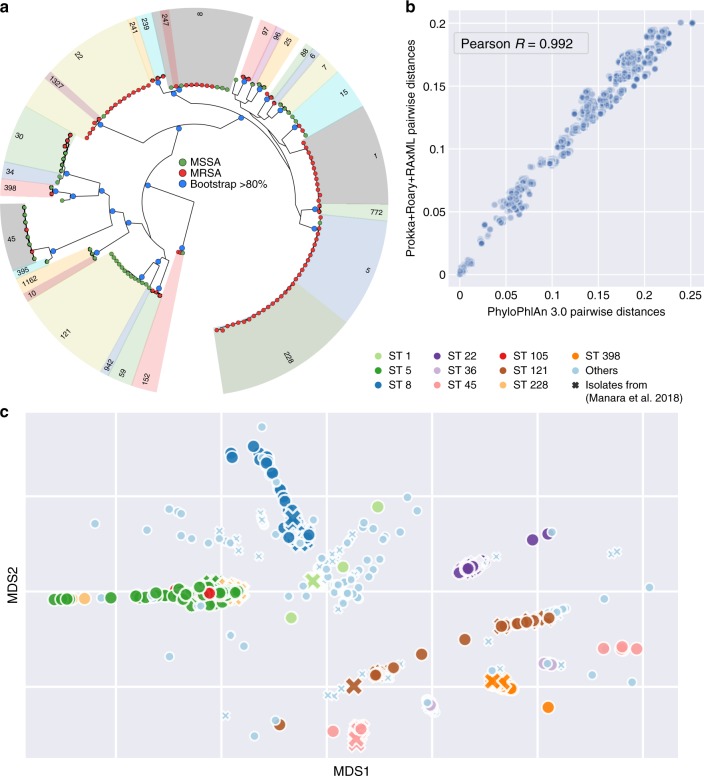
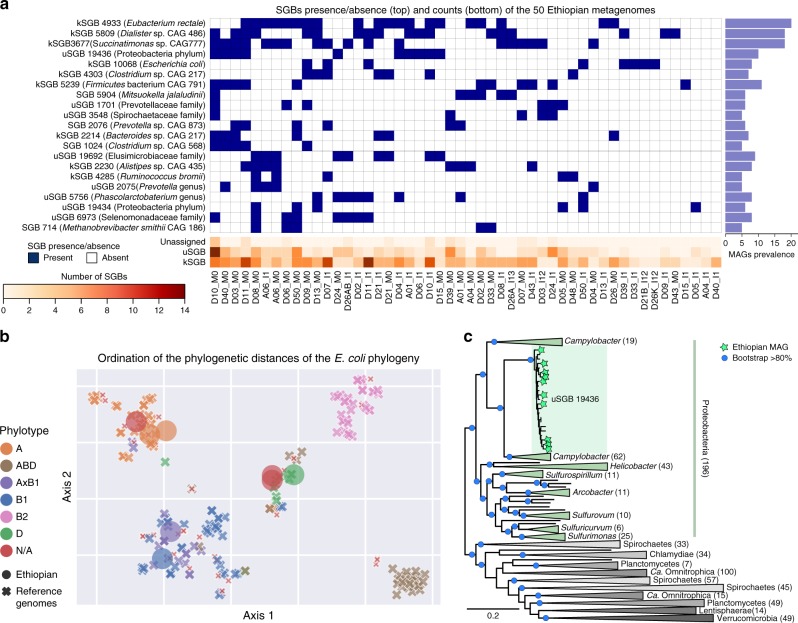
Microbial genomes are available at an ever-increasing pace, as cultivation and sequencing become cheaper and obtaining metagenome-assembled genomes (MAGs) becomes more effective. Phylogenetic placement methods to contextualize hundreds of thousands of genomes must thus be efficiently scalable and sensitive from closely related strains to divergent phyla. We present PhyloPhlAn 3.0, an accurate, rapid, and easy-to-use method for large-scale microbial genome characterization and phylogenetic analysis at multiple levels of resolution. PhyloPhlAn 3.0 can assign genomes from isolate sequencing or MAGs to species-level genome bins built from >230,000 publically available sequences. For individual clades of interest, it reconstructs strain-level phylogenies from among the closest species using clade-specific maximally informative markers. At the other extreme of resolution, it scales to large phylogenies comprising >17,000 microbial species. Examples including Staphylococcus aureus isolates, gut metagenomes, and meta-analyses demonstrate the ability of PhyloPhlAn 3.0 to support genomic and metagenomic analyses.
微生物基因组的获取速度越来越快,因为培养和测序变得更便宜,获得宏基因组组装基因组 (MAG) 变得更加有效。因此,必须有一种有效的、可扩展的、对从密切相关的菌株到不同门的菌株都敏感的系统发育定位方法来对数十万基因组进行上下文分析。我们提出了 PhyloPhlAn 3.0,这是一种用于大规模微生物基因组特征描述和在多个分辨率水平进行系统发育分析的准确、快速且易于使用的方法。PhyloPhlAn 3.0 可以将来自分离物测序或 MAG 的基因组分配到从 >230,000 个公开序列构建的物种级基因组 bin 中。对于感兴趣的个体分支,它使用特定分支的最大信息量标记从最接近的物种中重建菌株级系统发育。在分辨率的另一个极端,它可以扩展到包含 >17,000 个微生物物种的大型系统发育。包括金黄色葡萄球菌分离株、肠道宏基因组和荟萃分析在内的示例证明了 PhyloPhlAn 3.0 支持基因组和宏基因组分析的能力。