Medical Genomics Research Department, King Abdullah International Medical Research Center (KAIMRC), King Saud Bin Abdulaziz University for Health Sciences, King AbdulAziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Kingdom of Saudi Arabia.
Division of Genetics, Department of Pediatrics, King Abdullah Specialized Children's Hospital, King Saud Bin Abdulaziz University for Health Sciences, King Abdulaziz Medical City, Riyadh, Saudi Arabia.
Ann Clin Transl Neurol. 2020 Jun;7(6):956-964. doi: 10.1002/acn3.51059. Epub 2020 May 19.
RAP1GDS1 (RAP1, GTP-GDP dissociation stimulator 1), also known as SmgGDS, is a guanine nucleotide exchange factor (GEF) that regulates small GTPases, including, RHOA, RAC1, and KRAS. RAP1GDS1 was shown to be highly expressed in different tissue types including the brain. However, mutations in the RAP1GDS1 gene associated with human diseases have not previously been reported.
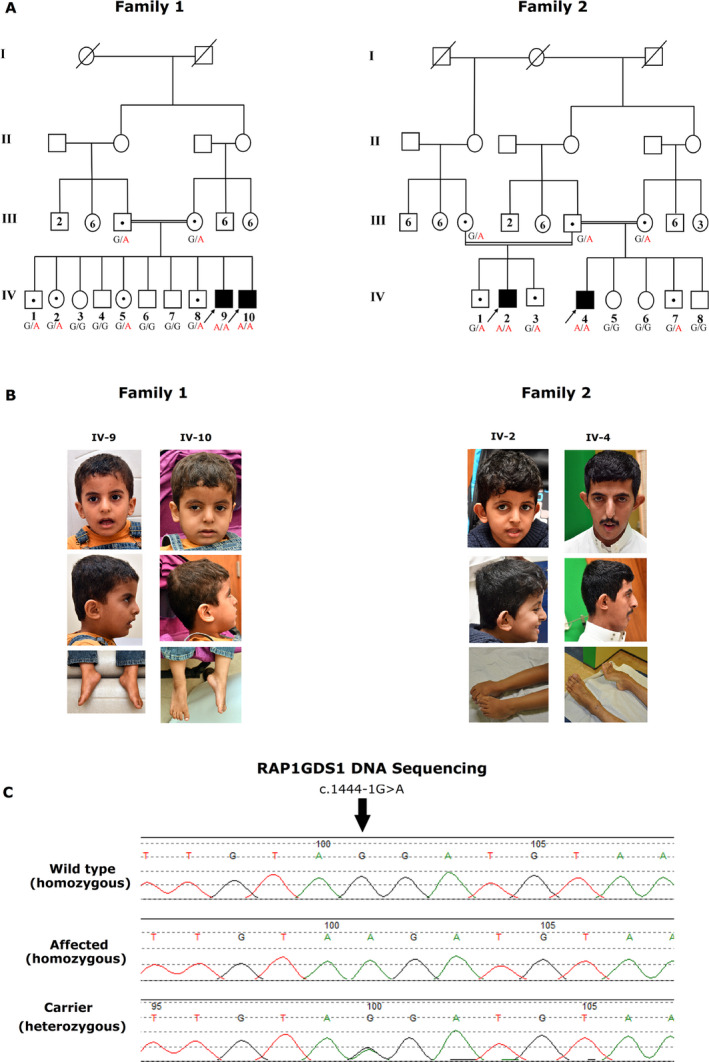
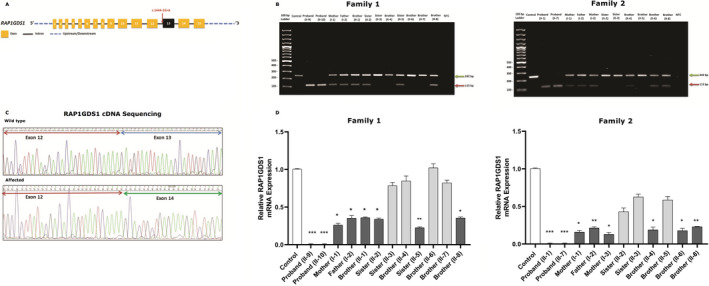
We report on four affected individuals, presenting intellectual disability, global developmental delay (GDD), and hypotonia. The probands' DNA was subjected to whole-genome sequencing, revealing a homozygous splice acceptor site mutation in the RAP1GDS1 gene (1444-1G > A). Sanger sequencing was performed to confirm the segregation of the variant in two Saudi families. The possible aberrant splicing in the patients' RNA was investigated using RT-PCR and changes in mRNA expression of the patients were confirmed using qRT-PCR.
The identified splice variant was found to segregate within the two families. RT-PCR showed that the mutation affected RAP1GDS1 gene splicing, resulting in the production of aberrant transcripts in the affected individuals. Quantitative gene expression analysis demonstrated that the RAP1GDS1 mRNA expression in all the probands was significantly decreased compared to that of the control, and Sanger sequencing of the probands' cDNA revealed skipping of exon 13, further strengthening the pathogenicity of this variant.
We are the first to report the mutation of the RAP1GDS1 gene as a potential cause of GDD and hypotonia. However, further investigations into the molecular mechanisms involved are required to confirm the role of RAP1GDS1 gene in causing GDD and hypotonia.
RAP1GDS1(RAP1,GTP-GDP 解离刺激因子 1),也称为 SmgGDS,是一种调节小 GTP 酶的鸟嘌呤核苷酸交换因子(GEF),包括 RHOA、RAC1 和 KRAS。RAP1GDS1 在不同的组织类型中高度表达,包括大脑。然而,以前没有报道过与人类疾病相关的 RAP1GDS1 基因突变。
我们报告了四个受影响的个体,表现为智力残疾、全面发育迟缓(GDD)和低张力。对先证者的 DNA 进行全基因组测序,发现 RAP1GDS1 基因的一个纯合剪接受体位点突变(1444-1G > A)。对两个沙特家族进行 Sanger 测序以确认该变体的分离。使用 RT-PCR 研究患者 RNA 中的可能异常剪接,并使用 qRT-PCR 证实患者的 mRNA 表达变化。
所鉴定的剪接变体在两个家族中分离。RT-PCR 显示该突变影响 RAP1GDS1 基因剪接,导致受影响个体产生异常转录本。定量基因表达分析表明,所有先证者的 RAP1GDS1 mRNA 表达均明显低于对照组,先证者 cDNA 的 Sanger 测序显示外显子 13 跳跃,进一步增强了该变体的致病性。
我们是第一个报道 RAP1GDS1 基因突变可能导致 GDD 和低张力的人。然而,需要进一步研究涉及的分子机制,以确认 RAP1GDS1 基因在导致 GDD 和低张力中的作用。