Medical Genomics Research Department, King Abdullah International Medical Research Center (KAIMRC), King Saud Bin Abdulaziz University for Health Sciences, Ministry of National Guard Health Affairs (MNGH), Riyadh, Saudi Arabia.
Department of Pathology and Laboratory Medicine, King Abdulaziz Medical City, Riyadh, Saudi Arabia.
Clin Genet. 2020 Dec;98(6):555-561. doi: 10.1111/cge.13842. Epub 2020 Sep 15.
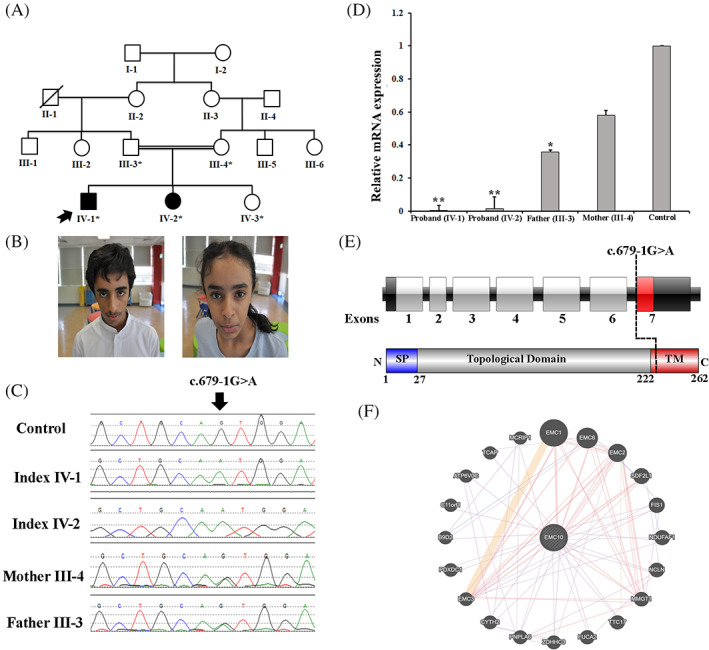
In recent years, several genes have been implicated in the variable disease presentation of global developmental delay (GDD) and intellectual disability (ID). The endoplasmic reticulum membrane protein complex (EMC) family is known to be involved in GDD and ID. Homozygous variants of EMC1 are associated with GDD, scoliosis, and cerebellar atrophy, indicating the relevance of this pathway for neurogenetic disorders. EMC10 is a bone marrow-derived angiogenic growth factor that plays an important role in infarct vascularization and promoting tissue repair. However, this gene has not been previously associated with human disease. Herein, we describe a Saudi family with two individuals segregating a recessive neurodevelopmental disorder. Both of the affected individuals showed mild ID, speech delay, and GDD. Whole-exome sequencing (WES) and Sanger sequencing were performed to identify candidate genes. Further, to elucidate the functional effects of the variant, quantitative real-time PCR (RT-qPCR)-based expression analysis was performed. WES revealed a homozygous splice acceptor site variant (c.679-1G>A) in EMC10 (chromosome 19q13.33) that segregated perfectly within the family. RT-qPCR showed a substantial decrease in the relative EMC10 gene expression in the patients, indicating the pathogenicity of the identified variant. For the first time in the literature, the EMC10 gene variant was associated with mild ID, speech delay, and GDD. Thus, this gene plays a key role in developmental milestones, with the potential to cause neurodevelopmental disorders in humans.
近年来,有几个基因被认为与全球发育迟缓(GDD)和智力障碍(ID)的疾病表现的多变性有关。内质网膜蛋白复合物(EMC)家族已知与 GDD 和 ID 有关。EMC1 的纯合变体与 GDD、脊柱侧凸和小脑萎缩有关,表明该途径与神经遗传疾病有关。EMC10 是一种骨髓来源的血管生成生长因子,在梗塞血管化和促进组织修复中发挥重要作用。然而,该基因以前与人类疾病无关。在此,我们描述了一个沙特家庭,其中有两个个体分离出隐性神经发育障碍。受影响的两个人都表现出轻度 ID、言语延迟和 GDD。进行全外显子组测序(WES)和 Sanger 测序以鉴定候选基因。此外,为了阐明变异的功能影响,进行了基于定量实时 PCR(RT-qPCR)的表达分析。WES 显示 EMC10(19q13.33 号染色体)中存在一个纯合剪接受体位点变异(c.679-1G>A),该变异在家族中完全分离。RT-qPCR 显示患者中相对 EMC10 基因表达显著降低,表明鉴定的变异具有致病性。这是文献中首次将 EMC10 基因变异与轻度 ID、言语延迟和 GDD 相关联。因此,该基因在发育里程碑中起着关键作用,有可能导致人类的神经发育障碍。