Department of Epidemiology, Biostatistics and Occupational Health, McGill University, Montreal, QC, Canada.
Lady Davis Institute for Medical Research, Montreal, QC, Canada.
Biometrics. 2021 Jun;77(2):424-438. doi: 10.1111/biom.13307. Epub 2020 Jun 5.
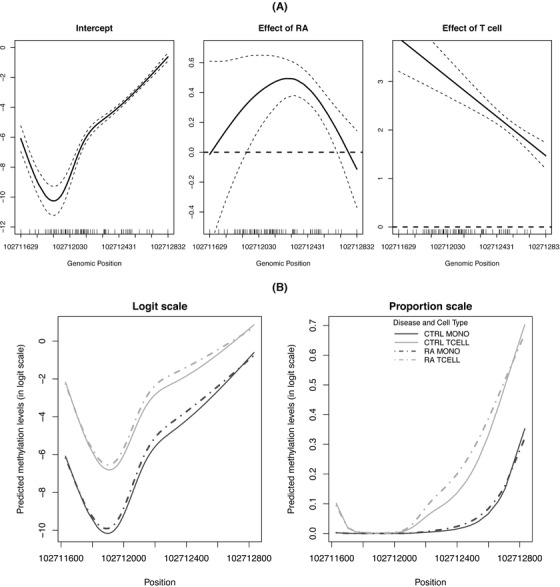
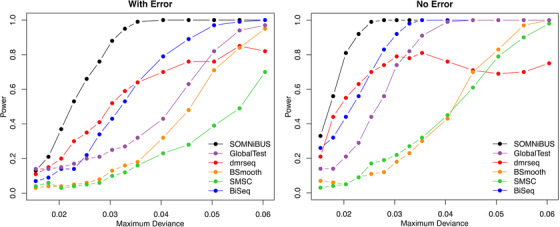
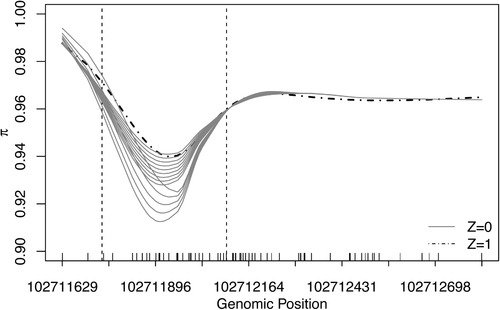
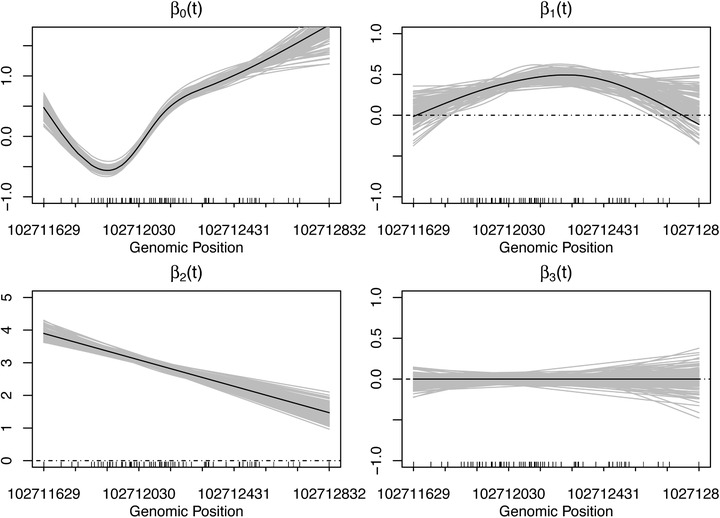
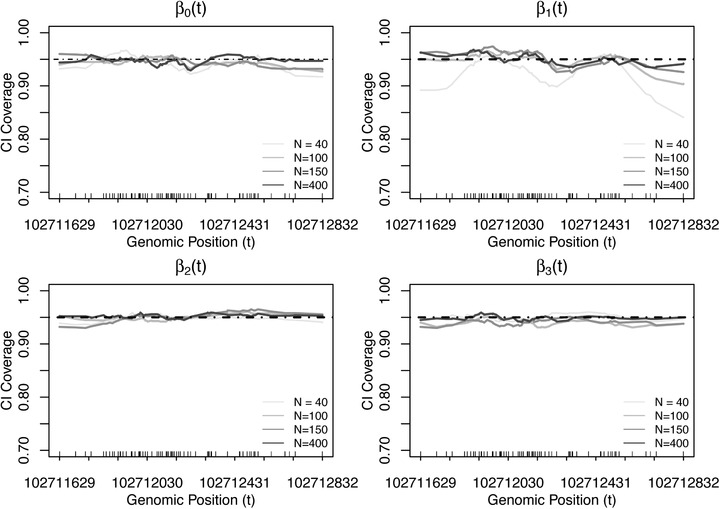
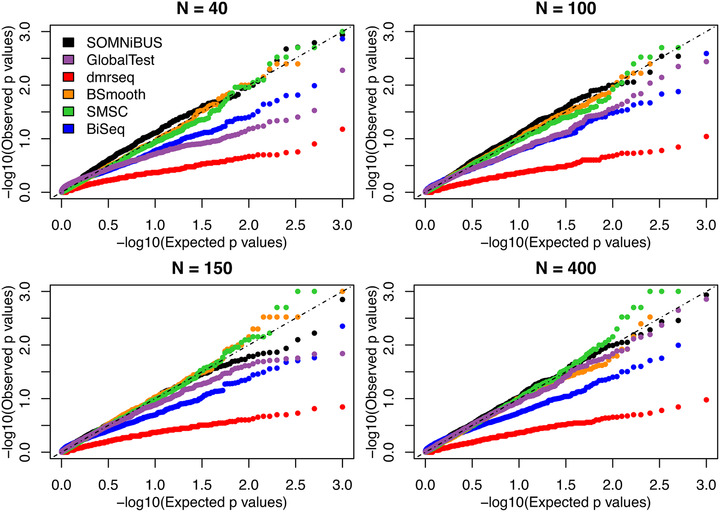
Identifying disease-associated changes in DNA methylation can help us gain a better understanding of disease etiology. Bisulfite sequencing allows the generation of high-throughput methylation profiles at single-base resolution of DNA. However, optimally modeling and analyzing these sparse and discrete sequencing data is still very challenging due to variable read depth, missing data patterns, long-range correlations, data errors, and confounding from cell type mixtures. We propose a regression-based hierarchical model that allows covariate effects to vary smoothly along genomic positions and we have built a specialized EM algorithm, which explicitly allows for experimental errors and cell type mixtures, to make inference about smooth covariate effects in the model. Simulations show that the proposed method provides accurate estimates of covariate effects and captures the major underlying methylation patterns with excellent power. We also apply our method to analyze data from rheumatoid arthritis patients and controls. The method has been implemented in R package SOMNiBUS.
鉴定 DNA 甲基化与疾病相关的变化可以帮助我们更好地了解疾病的病因。亚硫酸氢盐测序允许在 DNA 的单个碱基分辨率下生成高通量的甲基化图谱。然而,由于可变的读取深度、缺失数据模式、长程相关性、数据错误以及细胞类型混合物的混杂,最佳地建模和分析这些稀疏和离散的测序数据仍然是非常具有挑战性的。我们提出了一种基于回归的层次模型,允许协变量效应沿着基因组位置平滑变化,并且我们构建了一个专门的 EM 算法,该算法明确允许实验误差和细胞类型混合物,以便对模型中平滑的协变量效应进行推断。模拟表明,所提出的方法可以提供协变量效应的准确估计,并以优异的功效捕获主要的潜在甲基化模式。我们还将我们的方法应用于分析类风湿关节炎患者和对照的数据集。该方法已在 R 包 SOMNiBUS 中实现。