Statistics Department, Tel Aviv University, Tel Aviv, Israel.
Computer Science Department, Technion - Israel Institute of Technology, Haifa, Israel.
Bioinformatics. 2017 Jul 15;33(14):i325-i332. doi: 10.1093/bioinformatics/btx248.
Epigenome-wide association studies can provide novel insights into the regulation of genes involved in traits and diseases. The rapid emergence of bisulfite-sequencing technologies enables performing such genome-wide studies at the resolution of single nucleotides. However, analysis of data produced by bisulfite-sequencing poses statistical challenges owing to low and uneven sequencing depth, as well as the presence of confounding factors. The recently introduced Mixed model Association for Count data via data AUgmentation (MACAU) can address these challenges via a generalized linear mixed model when confounding can be encoded via a single variance component. However, MACAU cannot be used in the presence of multiple variance components. Additionally, MACAU uses a computationally expensive Markov Chain Monte Carlo (MCMC) procedure, which cannot directly approximate the model likelihood.
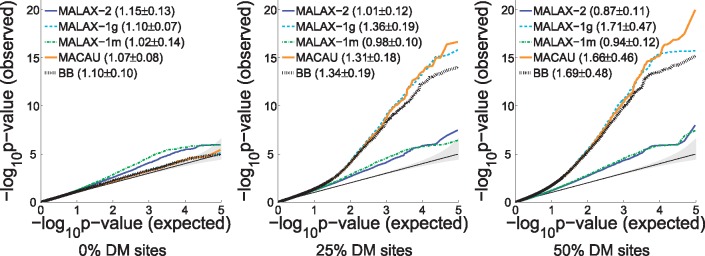
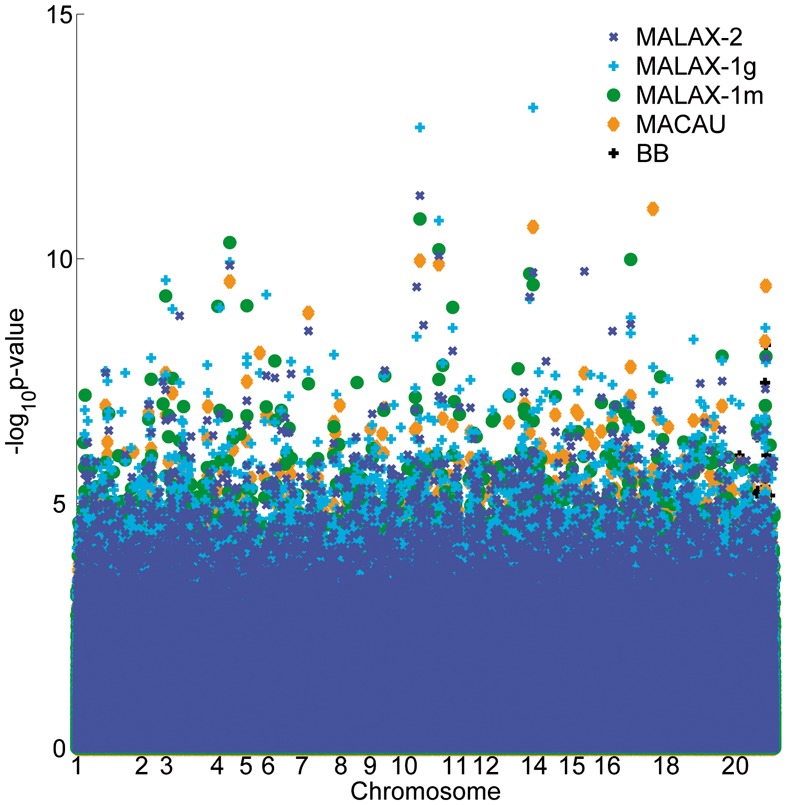
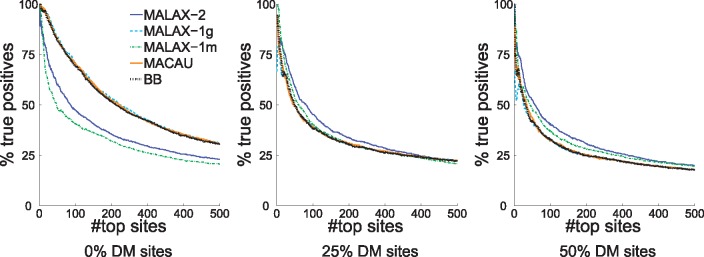
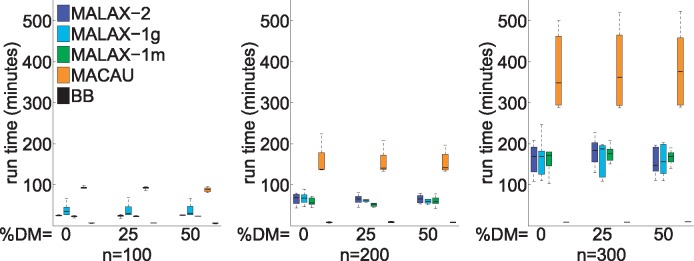
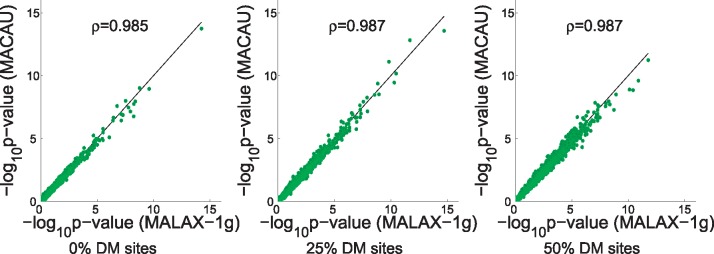
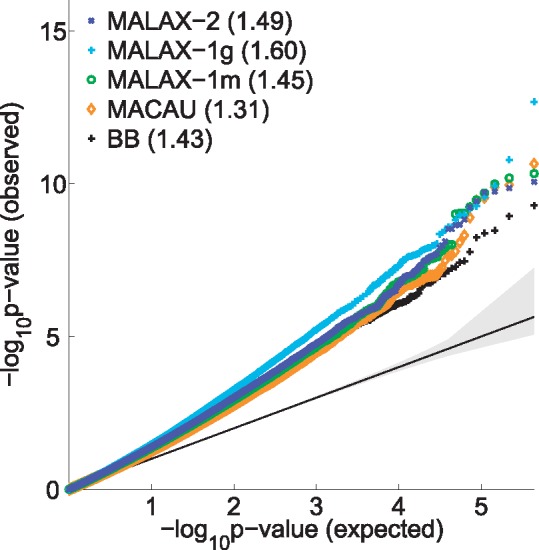
We present a new method, Mixed model Association via a Laplace ApproXimation (MALAX), that is more computationally efficient than MACAU and allows to model multiple variance components. MALAX uses a Laplace approximation rather than MCMC based approximations, which enables to directly approximate the model likelihood. Through an extensive analysis of simulated and real data, we demonstrate that MALAX successfully addresses statistical challenges introduced by bisulfite-sequencing while controlling for complex sources of confounding, and can be over 50% faster than the state of the art.
The full source code of MALAX is available at https://github.com/omerwe/MALAX .
omerw@cs.technion.ac.il or ehalperin@cs.ucla.edu.
Supplementary data are available at Bioinformatics online.
全基因组关联研究可以为涉及性状和疾病的基因调控提供新的见解。亚硫酸氢盐测序技术的快速出现使我们能够以单个核苷酸的分辨率进行此类全基因组研究。然而,由于测序深度低且不均匀,以及存在混杂因素,分析亚硫酸氢盐测序产生的数据存在统计学挑战。最近引入的通过数据增强进行计数数据混合模型关联(MACAU)可以通过广义线性混合模型来解决这些挑战,当混杂因素可以通过单个方差分量进行编码时。然而,当存在多个方差分量时,MACAU 无法使用。此外,MACAU 使用计算成本高的马尔可夫链蒙特卡罗(MCMC)过程,该过程不能直接逼近模型似然。
我们提出了一种新方法,即通过拉普拉斯逼近进行混合模型关联(MALAX),该方法比 MACAU 更具计算效率,并允许对多个方差分量进行建模。MALAX 使用拉普拉斯逼近而不是基于 MCMC 的逼近,这使得能够直接逼近模型似然。通过对模拟和真实数据的广泛分析,我们证明 MALAX 成功地解决了亚硫酸氢盐测序引入的统计挑战,同时控制了复杂的混杂来源,并且可以比最先进的方法快 50%以上。
MALAX 的完整源代码可在 https://github.com/omerwe/MALAX 上获得。
omerw@cs.technion.ac.il 或 ehalperin@cs.ucla.edu。
补充数据可在生物信息学在线获得。