Department of Physiology and Pathophysiology, The Fourth Military Medical University, Xi'an, 710032, China.
Department of Aerospace Medicine, The Fourth Military Medical University, Xi'an, 710032, China.
Cell Death Dis. 2020 May 21;11(5):388. doi: 10.1038/s41419-020-2605-y.
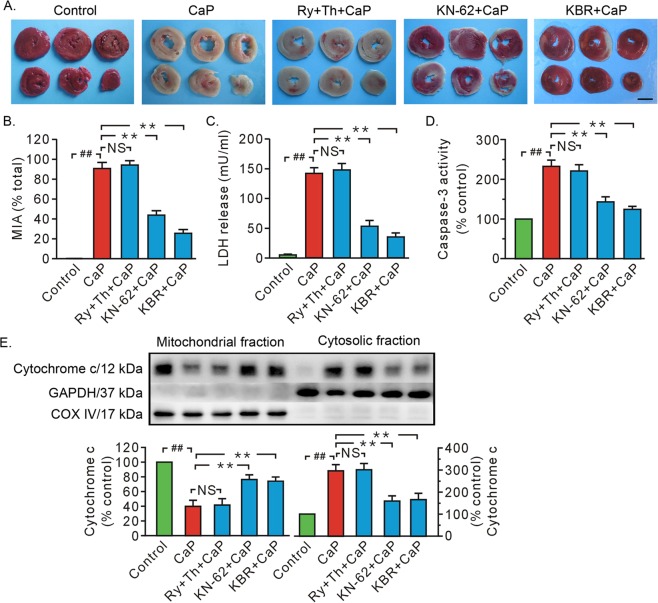
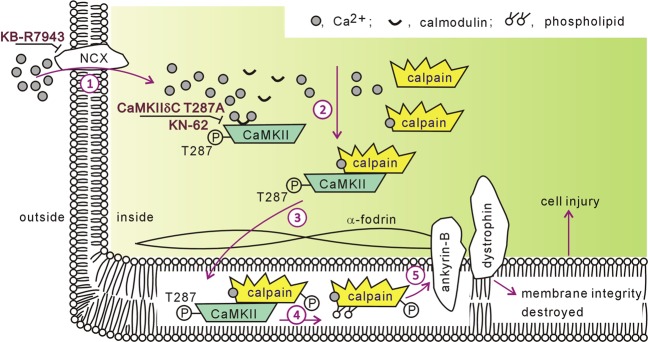
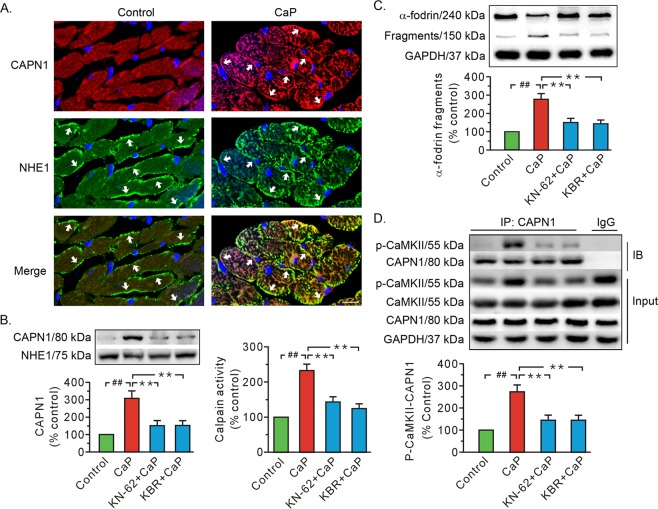
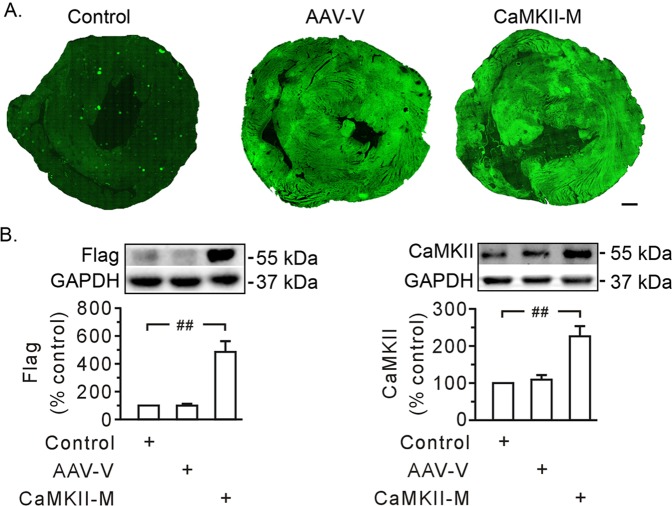
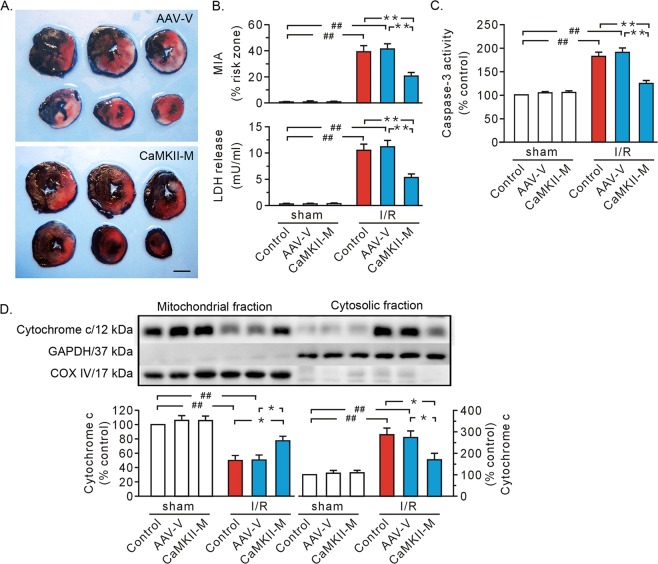
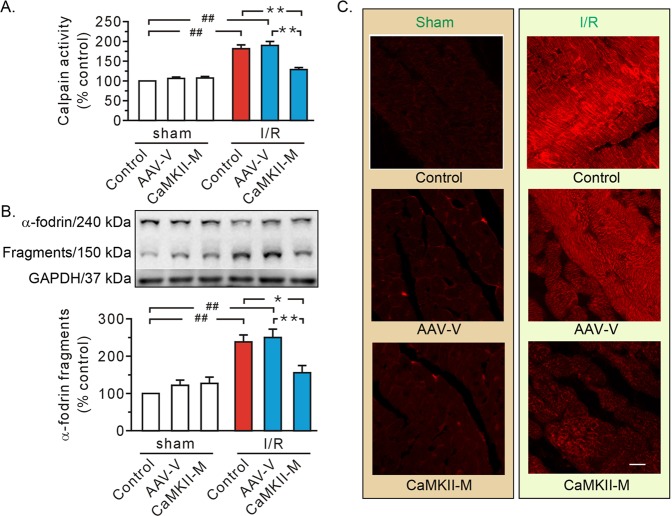
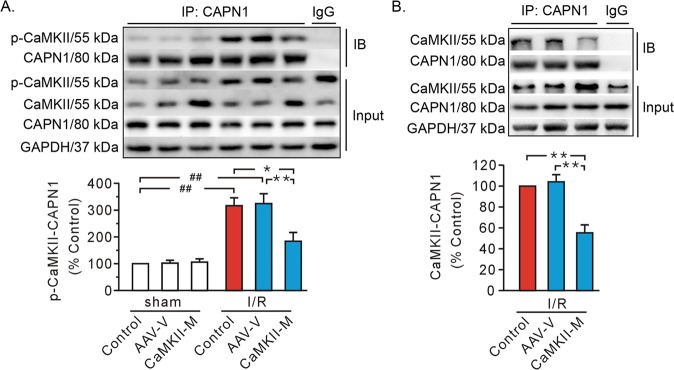
Previous studies indicated that Ca/calmodulin-dependent kinase II (CaMKII), a kinase involved in the modulation of ryanodine receptor activity, activates Ca-regulated protease μ-calpain to promote myocardial ischemia/reperfusion injury. This study was performed to explore the underlying mechanisms in CaMKII-induced calpain activation to better understand heart injury. To examine the Ca paradox and ischemia/reperfusion injury, isolated rat hearts were subjected to a Ca-free solution for 3 min, or left coronary artery occlusion for 40 min, prior to restoration of normal perfusion. Blockade of trans-sarcoplasmic reticulum Ca flux using ryanodine and thapsigargin failed to prevent Ca paradox-induced heart injury. In contrast, the Ca paradox increased CaMKII auto-phosphorylation at Thr, while the CaMKII inhibitor KN-62 and the Na/Ca exchanger inhibitor KB-R7943 alleviated heart injury and calpain activity. Intriguingly, the binding of μ-calpain large subunit calpain-1 (CAPN1) to phospho-CaMKII was blunted by both inhibitors. Thus, a Ca leak via the ryanodine receptor is not an essential element in CaMKII-elicited calpain activation. In hearts receiving vector injection, ischemia/reperfusion caused elevated calpain activity and α-fodrin degradation, along with membrane integrity damage, similar to the effects noted in control hearts. Importantly, all these alterations were diminished with delivery of adeno-associated virus expressing mutant CaMKIIδC T287A. Ischemia/reperfusion increased CaMKII auto-phosphorylation and binding of CAPN1 to phospho-CaMKII, and facilitated the translocation of phospho-CaMKII and CAPN1 to the plasma membrane, all of which were reversed by injecting CaMKII mutant. Furthermore, the relocation capacity and the interaction of CaMKII with CAPN1 appeared to be dependent upon CaMKII autophosphorylation, as its mutant delivery increased the level of CaMKII, but did not increase membrane content of CaMKII and CAPN1, or their interactions. Together, CaMKII/calpain interaction represents a new avenue for mediating myocardial ischemia/reperfusion injury, and CaMKII likely serves as both a kinase and a carrier, thereby promoting calpain membrane translocation and activation.
先前的研究表明,钙/钙调蛋白依赖性激酶 II(CaMKII)是一种参与调节兰尼碱受体活性的激酶,它可激活钙调节蛋白酶μ-钙蛋白酶,从而促进心肌缺血/再灌注损伤。本研究旨在探讨 CaMKII 诱导钙蛋白酶激活的潜在机制,以便更好地理解心脏损伤。为了研究钙反常和缺血/再灌注损伤,分离的大鼠心脏先在无钙溶液中处理 3 分钟,或左冠状动脉闭塞 40 分钟,然后再恢复正常灌注。使用兰尼碱和 thapsigargin 阻断跨肌浆网钙流未能阻止钙反常引起的心脏损伤。相反,钙反常增加了 CaMKII 自身 Thr 磷酸化,而 CaMKII 抑制剂 KN-62 和 Na/Ca 交换体抑制剂 KB-R7943 减轻了心脏损伤和钙蛋白酶活性。有趣的是,两种抑制剂均减弱了 μ-钙蛋白酶大亚基钙蛋白酶-1(CAPN1)与磷酸化-CaMKII 的结合。因此,兰尼碱受体的钙漏不是 CaMKII 引发钙蛋白酶激活的必要因素。在接受载体注射的心脏中,缺血/再灌注导致钙蛋白酶活性升高和α-辅肌动蛋白降解,以及膜完整性损伤,与对照心脏中的作用相似。重要的是,用表达突变型 CaMKIIδC T287A 的腺相关病毒表达载体转染后,所有这些改变均减轻。缺血/再灌注增加了 CaMKII 的自身磷酸化和 CAPN1 与磷酸化-CaMKII 的结合,并促进了磷酸化-CaMKII 和 CAPN1 向质膜的易位,这些改变均可被 CaMKII 突变体逆转。此外,CaMKII 与 CAPN1 的重定位能力及其相互作用似乎依赖于 CaMKII 的自身磷酸化,因为其突变体的传递增加了 CaMKII 的水平,但并未增加质膜中 CaMKII 和 CAPN1 的水平,或它们之间的相互作用。总之,CaMKII/钙蛋白酶相互作用为介导心肌缺血/再灌注损伤提供了一个新途径,CaMKII 可能既是激酶又是载体,从而促进钙蛋白酶向质膜的易位和激活。