Dorothy M. Davis Heart and Lung Research Institute, College of Medicine, The Ohio State University, Columbus, OH, 43210, USA.
Department of Physiology and Cell Biology, College of Medicine, The Ohio State University, Columbus, OH, 43210, USA.
Basic Res Cardiol. 2020 May 22;115(4):38. doi: 10.1007/s00395-020-0797-z.
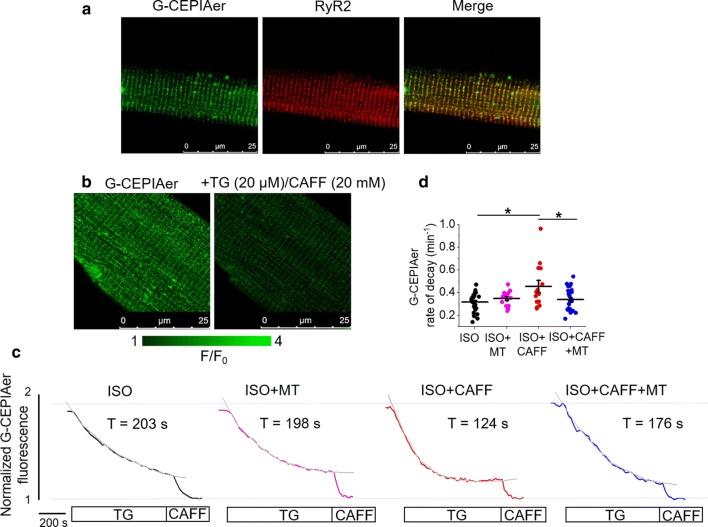
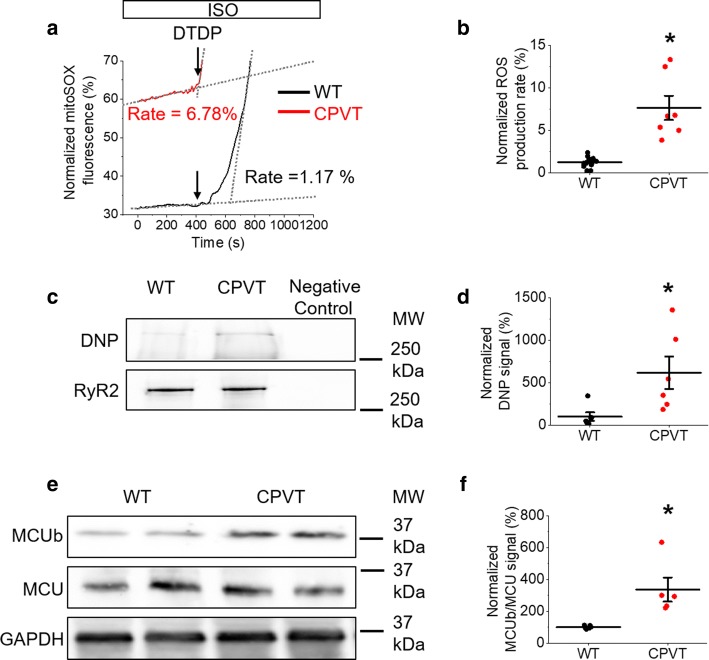
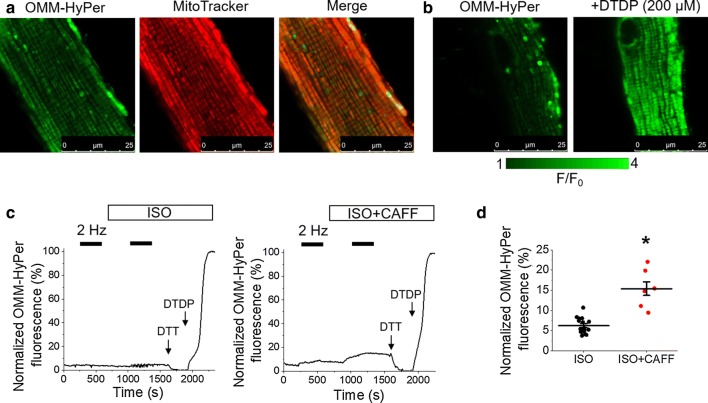
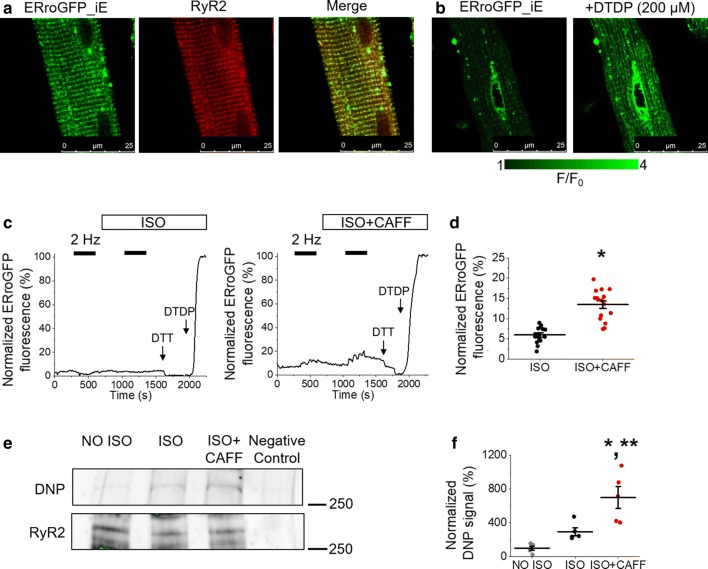
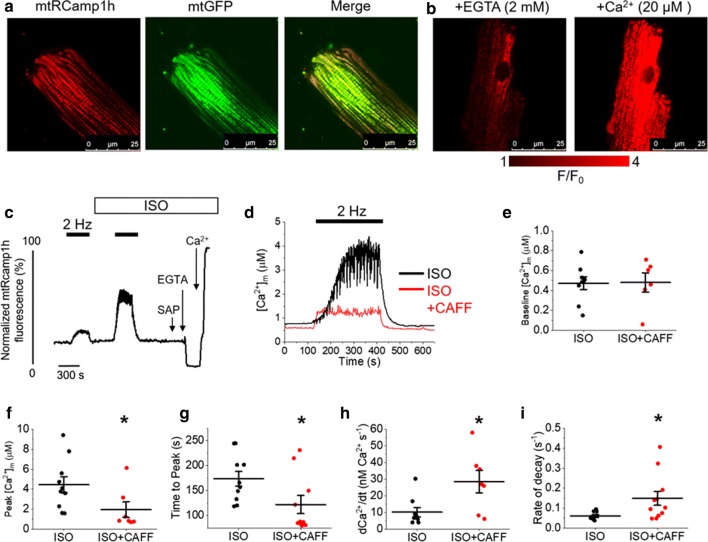
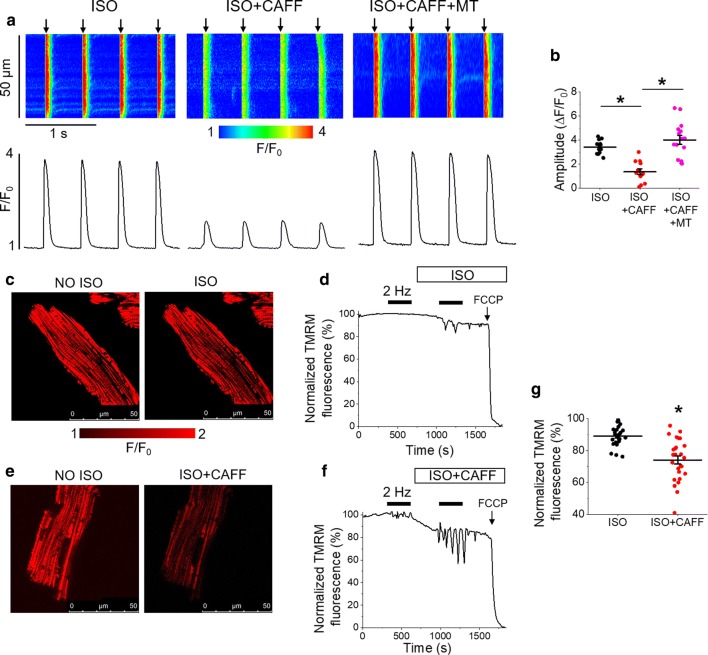
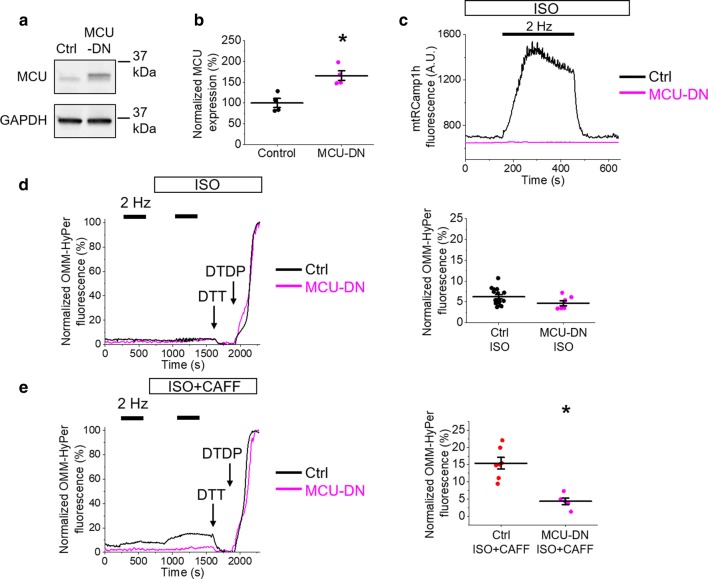
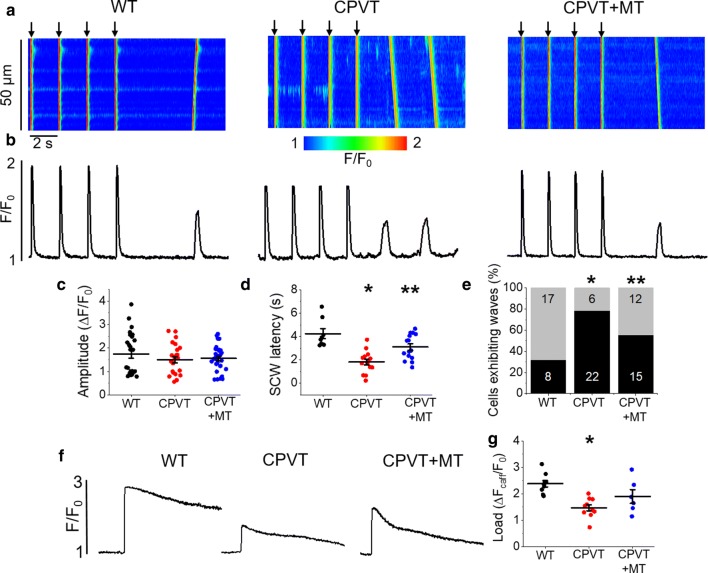
Cardiac disease is associated with deleterious emission of mitochondrial reactive oxygen species (mito-ROS), as well as enhanced oxidation and activity of the sarcoplasmic reticulum (SR) Ca release channel, the ryanodine receptor (RyR2). The transfer of Ca from the SR via RyR2 to mitochondria is thought to play a key role in matching increased metabolic demand during stress. In this study, we investigated whether augmented RyR2 activity results in self-imposed exacerbation of SR Ca leak, via altered SR-mitochondrial Ca transfer and elevated mito-ROS emission. Fluorescent indicators and spatially restricted genetic ROS probes revealed that both pharmacologically and genetically enhanced RyR2 activity, in ventricular myocytes from rats and catecholaminergic polymorphic ventricular tachycardia (CPVT) mice, respectively, resulted in increased ROS emission under β-adrenergic stimulation. Expression of mitochondrial Ca probe mtRCamp1h revealed diminished net mitochondrial [Ca] with enhanced SR Ca leak, accompanied by depolarization of the mitochondrial matrix. While this may serve as a protective mechanism to prevent mitochondrial Ca overload, protection is not complete and enhanced mito-ROS emission resulted in oxidation of RyR2, further amplifying proarrhythmic SR Ca release. Importantly, the effects of augmented RyR2 activity could be attenuated by mitochondrial ROS scavenging, and experiments with dominant-negative paralogs of the mitochondrial Ca uniporter (MCU) supported the hypothesis that SR-mitochondria Ca transfer is essential for the increase in mito-ROS. We conclude that in a process whereby leak begets leak, augmented RyR2 activity modulates mitochondrial Ca handling, promoting mito-ROS emission and driving further channel activity in a proarrhythmic feedback cycle in the diseased heart.
心脏疾病与线粒体活性氧物质(mito-ROS)的有害排放以及肌浆网(SR)Ca 释放通道,即兰尼碱受体(RyR2)的氧化和活性增强有关。SR 通过 RyR2 向线粒体转移的 Ca 被认为在应激时匹配增加的代谢需求中发挥关键作用。在这项研究中,我们研究了增强的 RyR2 活性是否通过改变 SR-线粒体 Ca 转移和升高的 mito-ROS 排放而导致自身加重 SR Ca 渗漏。荧光指示剂和空间限制的遗传 ROS 探针显示,在来自大鼠的心室肌细胞和儿茶酚胺多形性室性心动过速(CPVT)小鼠的分别药理学和遗传学增强的 RyR2 活性下,β-肾上腺素能刺激下均导致 ROS 发射增加。表达线粒体 Ca 探针 mtRCamp1h 显示,增强的 SR Ca 渗漏伴随着线粒体基质去极化,导致净线粒体[Ca]减少。虽然这可能是一种防止线粒体 Ca 过载的保护机制,但保护并不完全,增强的 mito-ROS 发射导致 RyR2 氧化,进一步放大促心律失常的 SR Ca 释放。重要的是,通过线粒体 ROS 清除可以减弱增强的 RyR2 活性的作用,并且使用线粒体 Ca 单向转运蛋白(MCU)的显性负效平行物的实验支持了这样的假设,即 SR-线粒体 Ca 转移对于增加 mito-ROS 是必需的。我们得出结论,在一个渗漏导致渗漏的过程中,增强的 RyR2 活性调节线粒体 Ca 处理,促进 mito-ROS 发射,并在患病心脏中驱动进一步的通道活性,形成促心律失常的反馈循环。