Department of Biological Sciences, Mississippi State University, 295 Lee Blvd, Starkville, Mississippi, 39762, USA.
epartment of Orthopedics, Center for Musculoskeletal Research, University of Rochester, 601 Elmwood Ave, Rochester, New York 14624, USA.
Cardiovasc Res. 2022 Oct 21;118(13):2819-2832. doi: 10.1093/cvr/cvab324.
Diastolic Ca release (DCR) from sarcoplasmic reticulum (SR) Ca release channel ryanodine receptor (RyR2) has been linked to multiple cardiac pathologies, but its exact role in shaping divergent cardiac pathologies remains unclear. We hypothesize that the SR-mitochondria interplay contributes to disease phenotypes by shaping Ca signalling.
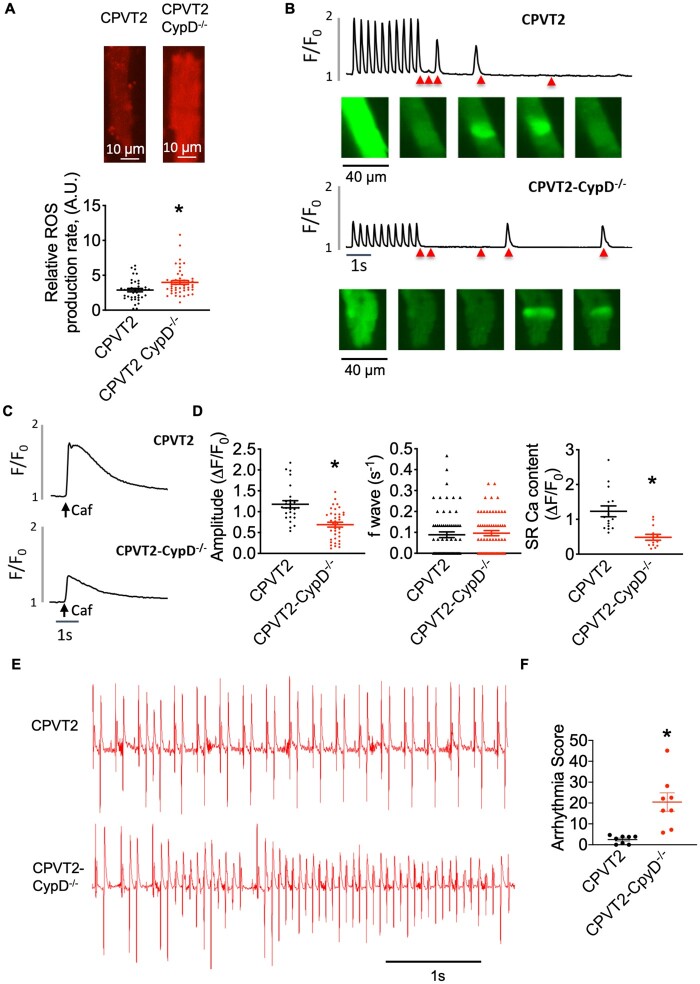
A genetic model of catecholaminergic polymorphic ventricular tachycardia (CPVT2 model of CASQ2 knockout) and a pre-diabetic cardiomyopathy model of fructose-fed mice (FFD), both marked by DCR, are employed in this study. Mitochondria Ca (mCa) is modulated by pharmacologically targeting mitochondria Ca uniporter (MCU) or permeability transition pore (mPTP), mCa uptake, and extrusion mechanisms, respectively. An MCU activator abolished Ca waves in CPVT2 but exacerbated waves in FFD cells. Mechanistically this is ascribed to mitochondria's function as a Ca buffer or source of reactive oxygen species (mtROS) to exacerbate RyR2 functionality, respectively. Enhancing mCa uptake reduced and elevated mtROS production in CPVT2 and FFD, respectively. In CPVT2, mitochondria took up more Ca in permeabilized cells, and had higher level of mCa content in intact cells vs. FFD. Conditional ablation of MCU in the CPVT2 model caused lethality and cardiac remodelling, but reduced arrhythmias in the FFD model. In parallel, CPVT2 mitochondria also employ up-regulated mPTP-mediated Ca efflux to avoid mCa overload, as seen by elevated incidence of MitoWinks (an indicator of mPTP-mediated Ca efflux) vs. FFD. Both pharmacological and genetic inhibition of mPTP promoted mtROS production and exacerbation of myocyte Ca handling in CPVT2. Further, genetic inhibition of mPTP exacerbated arrhythmias in CPVT2.
In contrast to FFD, which is more susceptible to mtROS-dependent RyR2 leak, in CPVT2 mitochondria buffer SR-derived DCR to mitigate Ca-dependent pathological remodelling and rely on mPTP-mediated Ca efflux to avoid mCa overload. SR-mitochondria interplay contributes to the divergent pathologies by disparately shaping intracellular Ca signalling.
肌浆网 Ca 释放通道兰尼碱受体(RyR2)介导的舒张期 Ca 释放(DCR)与多种心脏病理有关,但它在塑造不同心脏病理中的确切作用仍不清楚。我们假设 SR-线粒体相互作用通过调节 Ca 信号来影响疾病表型。
本研究采用儿茶酚胺多形性室性心动过速(CPVT2 型 CASQ2 敲除模型)和果糖喂养小鼠(FFD)的糖尿病前期心肌病模型,这两种模型均表现出 DCR。通过药理学靶向线粒体 Ca 单向转运体(MCU)或通透性转换孔(mPTP)、mCa 摄取和外排机制分别调节线粒体 Ca(mCa)。MCU 激活剂消除了 CPVT2 中的 Ca 波,但加剧了 FFD 细胞中的 Ca 波。从机制上讲,这归因于线粒体作为 Ca 缓冲剂或活性氧物质(mtROS)源的功能,分别增强 RyR2 功能。增强 mCa 摄取减少了 CPVT2 和 FFD 中的 mtROS 产生,反之亦然。在 CPVT2 中,与 FFD 相比,线粒体在通透细胞中摄取更多的 Ca,并且在完整细胞中具有更高的 mCa 含量。在 CPVT2 模型中条件性敲除 MCU 导致致死性和心脏重构,但减少了 FFD 模型中的心律失常。同时,CPVT2 线粒体还采用上调的 mPTP 介导的 Ca 外排来避免 mCa 过载,这可以通过增加 MitoWinks(一种 mPTP 介导的 Ca 外排的指标)的发生率与 FFD 相比看出。mPTP 的药理学和基因抑制均促进 CPVT2 中的 mtROS 产生和肌细胞 Ca 处理的恶化。此外,mPTP 的基因抑制加剧了 CPVT2 中的心律失常。
与更易受 mtROS 依赖性 RyR2 渗漏影响的 FFD 相反,在 CPVT2 中,线粒体缓冲 SR 来源的 DCR 以减轻 Ca 依赖性病理性重构,并依赖 mPTP 介导的 Ca 外排来避免 mCa 过载。SR-线粒体相互作用通过不同的方式调节细胞内 Ca 信号来影响不同的病理。