Neuroscience Program, Center for Applied Medical Research (CIMA), University of Navarra, 31008, Pamplona, Spain.
Gene Therapy and Regulation of Gene Expression Program, Center for Applied Medical Research (CIMA), University of Navarra, 31008, Pamplona, Spain.
Cell Death Dis. 2020 May 26;11(5):397. doi: 10.1038/s41419-020-2601-2.
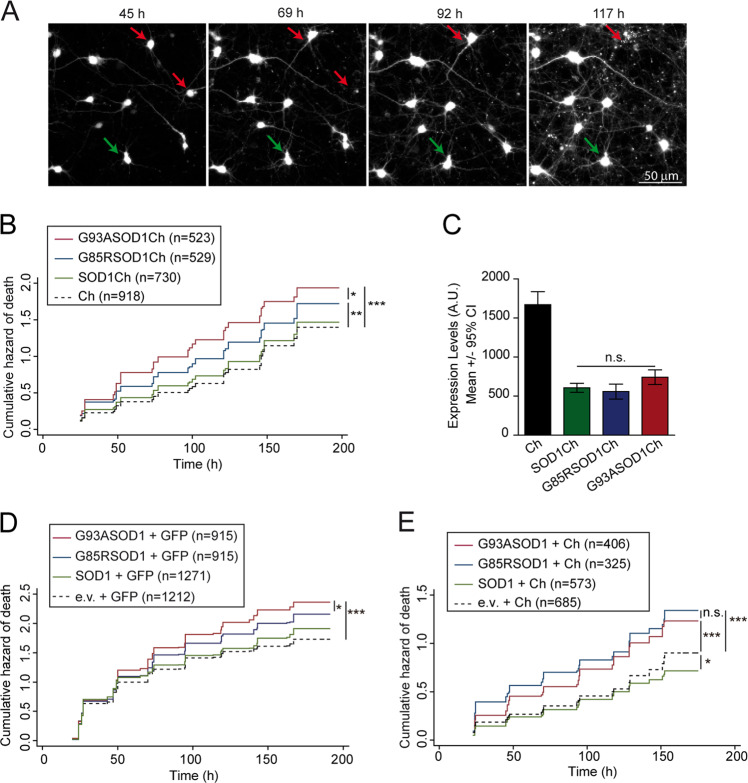
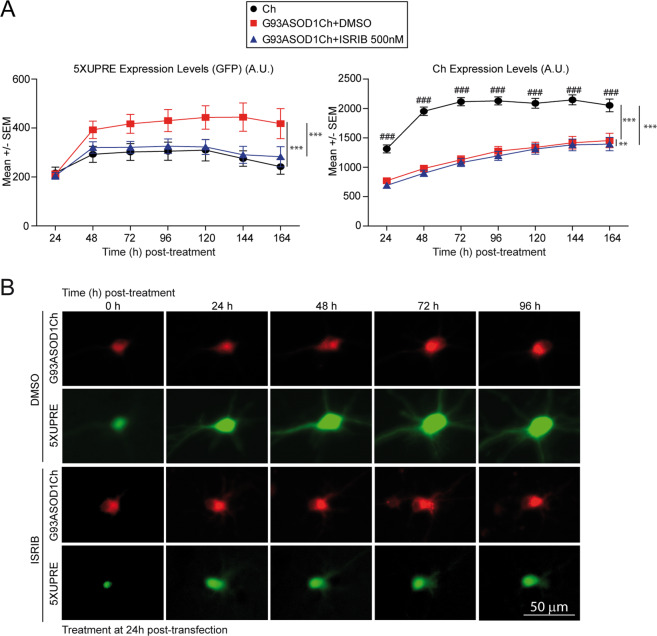
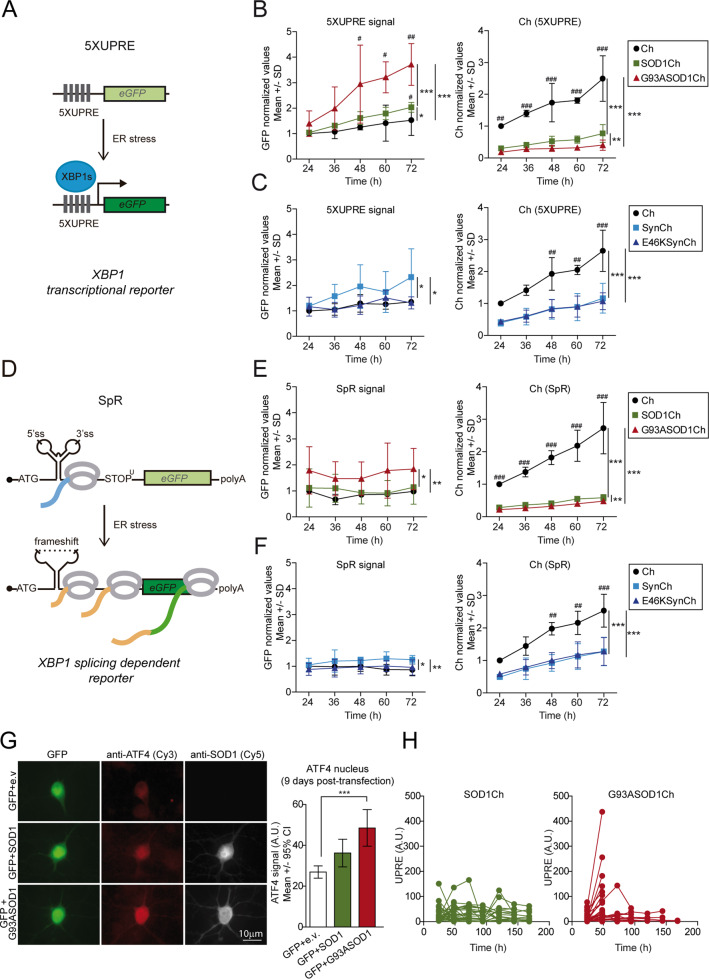
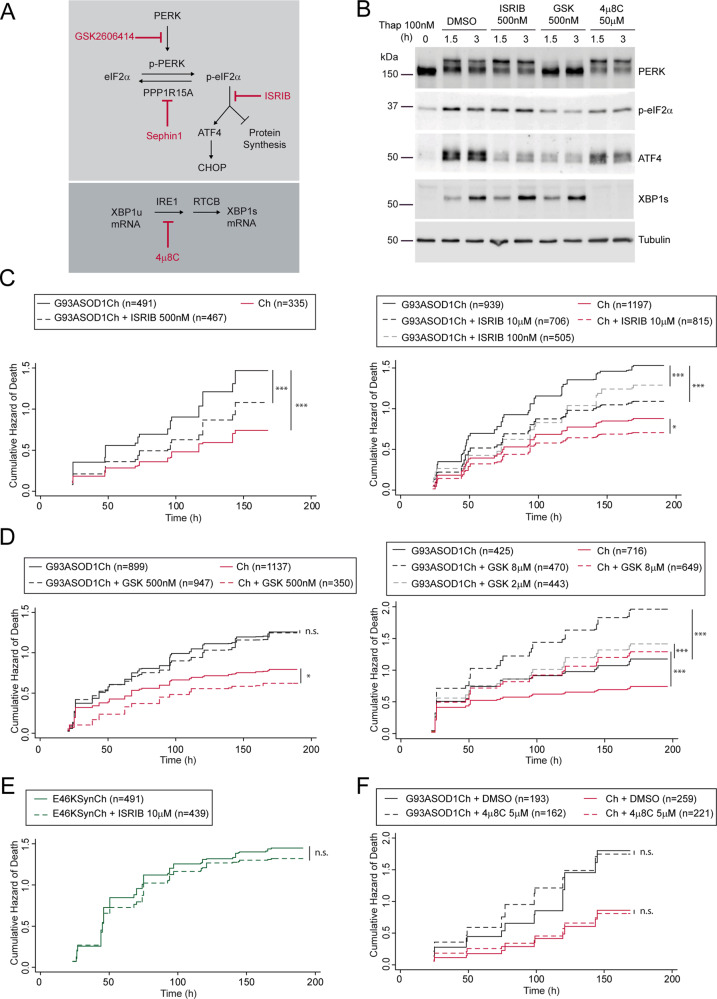
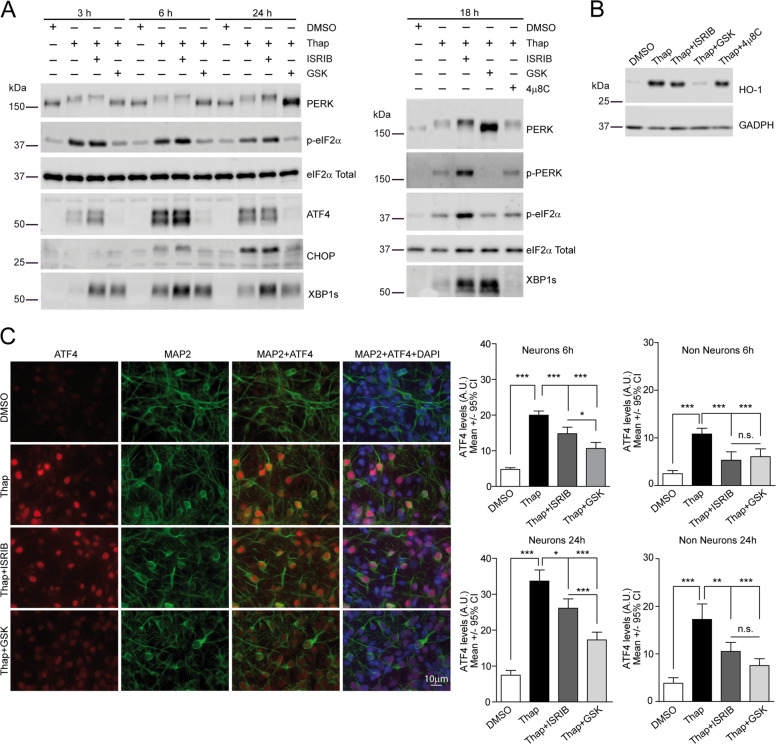
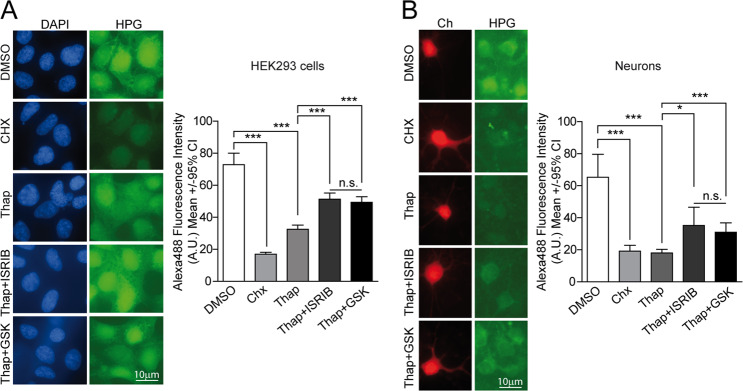
Loss of protein folding homeostasis features many of the most prevalent neurodegenerative disorders. As coping mechanism to folding stress within the endoplasmic reticulum (ER), the unfolded protein response (UPR) comprises a set of signaling mechanisms that initiate a gene expression program to restore proteostasis, or when stress is chronic or overwhelming promote neuronal death. This fate-defining capacity of the UPR has been proposed to play a key role in amyotrophic lateral sclerosis (ALS). However, the several genetic or pharmacological attempts to explore the therapeutic potential of UPR modulation have produced conflicting observations. In order to establish the precise relationship between UPR signaling and neuronal death in ALS, we have developed a neuronal model where the toxicity of a familial ALS-causing allele (mutant G93A SOD1) and UPR activation can be longitudinally monitored in single neurons over the process of neurodegeneration by automated microscopy. Using fluorescent UPR reporters we established the temporal and causal relationship between UPR and neuronal death by Cox regression models. Pharmacological inhibition of discrete UPR processes allowed us to establish the contribution of PERK (PKR-like ER kinase) and IRE1 (inositol-requiring enzyme-1) mechanisms to neuronal fate. Importantly, inhibition of PERK signaling with its downstream inhibitor ISRIB, but not with the direct PERK kinase inhibitor GSK2606414, significantly enhanced the survival of G93A SOD1-expressing neurons. Characterization of the inhibitory properties of both drugs under ER stress revealed that in neurons (but not in glial cells) ISRIB overruled only part of the translational program imposed by PERK, relieving the general inhibition of translation, but maintaining the privileged translation of ATF4 (activating transcription factor 4) messenger RNA. Surprisingly, the fine-tuning of the PERK output in G93A SOD1-expressing neurons led to a reduction of IRE1-dependent signaling. Together, our findings identify ISRIB-mediated translational reprogramming as a new potential ALS therapy.
蛋白质折叠稳态的丧失是许多最常见的神经退行性疾病的特征。作为内质网(ER)内折叠应激的应对机制,未折叠蛋白反应(UPR)包括一组信号机制,这些机制启动基因表达程序以恢复蛋白质稳态,或者当应激是慢性的或无法承受时,促进神经元死亡。UPR 的这种决定命运的能力被认为在肌萎缩侧索硬化症(ALS)中发挥关键作用。然而,几种遗传或药理学尝试探索 UPR 调节的治疗潜力产生了相互矛盾的观察结果。为了确定 UPR 信号与 ALS 中神经元死亡之间的确切关系,我们开发了一种神经元模型,在该模型中,可以通过自动显微镜在单个神经元中对家族性 ALS 致病等位基因(突变 G93A SOD1)的毒性和 UPR 激活进行纵向监测,以监测神经变性过程。使用荧光 UPR 报告基因,我们通过 Cox 回归模型建立了 UPR 和神经元死亡之间的时间和因果关系。对离散 UPR 过程的药理学抑制使我们能够确定 PERK(PKR 样 ER 激酶)和 IRE1(需要肌醇的酶-1)机制对神经元命运的贡献。重要的是,用其下游抑制剂 ISRIB 而不是直接的 PERK 激酶抑制剂 GSK2606414 抑制 PERK 信号,可显著提高表达 G93A SOD1 的神经元的存活率。在 ER 应激下,两种药物的抑制特性的表征表明,在神经元(而不是神经胶质细胞)中,ISRIB 仅部分推翻了 PERK 施加的翻译程序,从而缓解了翻译的普遍抑制,但维持了 ATF4(激活转录因子 4)信使 RNA 的特权翻译。令人惊讶的是,在表达 G93A SOD1 的神经元中精细调整 PERK 输出会导致 IRE1 依赖性信号的减少。总的来说,我们的研究结果确定了 ISRIB 介导的翻译重编程作为一种新的潜在 ALS 治疗方法。