Brief Bioinform. 2021 May 20;22(3). doi: 10.1093/bib/bbaa045.
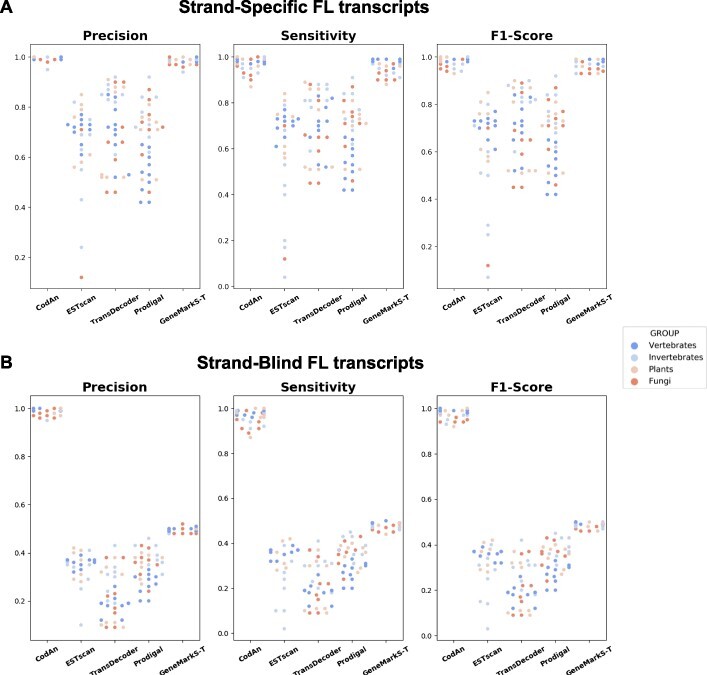
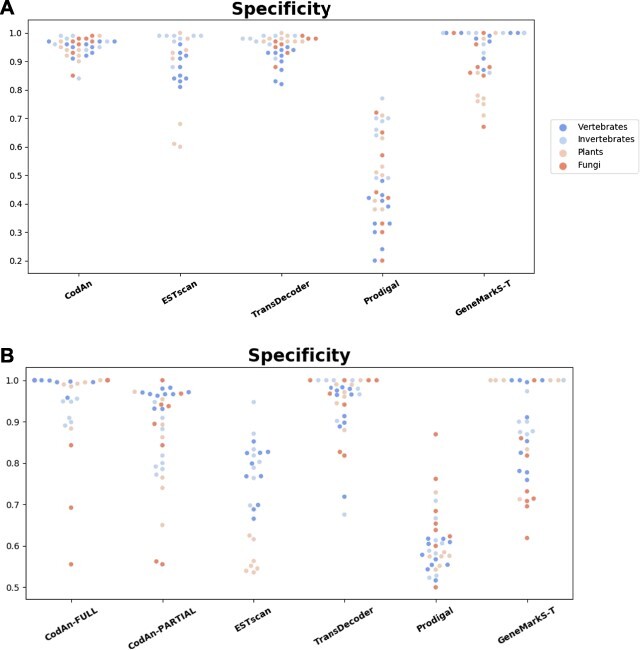
Characterization of the coding sequences (CDSs) is an essential step in transcriptome annotation. Incorrect identification of CDSs can lead to the prediction of non-existent proteins that can eventually compromise knowledge if databases are populated with similar incorrect predictions made in different genomes. Also, the correct identification of CDSs is important for the characterization of the untranslated regions (UTRs), which are known to be important regulators of the mRNA translation process. Considering this, we present CodAn (Coding sequence Annotator), a new approach to predict confident CDS and UTR regions in full or partial transcriptome sequences in eukaryote species.
Our analysis revealed that CodAn performs confident predictions on full-length and partial transcripts with the strand sense of the CDS known or unknown. The comparative analysis showed that CodAn presents better overall performance than other approaches, mainly when considering the correct identification of the full CDS (i.e. correct identification of the start and stop codons). In this sense, CodAn is the best tool to be used in projects involving transcriptomic data.
CodAn is freely available at https://github.com/pedronachtigall/CodAn.
Supplementary data are available at Briefings in Bioinformatics online.
对编码序列(CDS)进行特征描述是转录组注释的关键步骤。如果在不同的基因组中做出了类似的错误预测并将其填充到数据库中,那么错误识别 CDS 可能会导致预测出不存在的蛋白质,最终会影响知识的完整性。此外,正确识别 CDS 对于非翻译区(UTR)的特征描述也很重要,因为 UTR 已知是 mRNA 翻译过程的重要调节因子。考虑到这一点,我们提出了 CodAn(编码序列注释器),这是一种用于预测真核生物全长或部分转录组序列中置信 CDS 和 UTR 区的新方法。
我们的分析表明,CodAn 可以在已知或未知 CDS 链的全长和部分转录本上进行可靠的预测。比较分析表明,CodAn 比其他方法具有更好的整体性能,尤其是在正确识别全长 CDS 方面(即正确识别起始和终止密码子)。从这个意义上说,CodAn 是涉及转录组数据项目的最佳工具。
CodAn 可在 https://github.com/pedronachtigall/CodAn 上免费获得。
补充材料可在Briefings in Bioinformatics 在线获取。