Department of Pharmacology and Toxicology, University of Mississippi Medical Center, Jackson, MS, USA.
Department of General Surgery, Third Xiangya Hospital, Central South University, Changsha, China.
J Cell Mol Med. 2020 Jul;24(14):8057-8068. doi: 10.1111/jcmm.15437. Epub 2020 May 28.
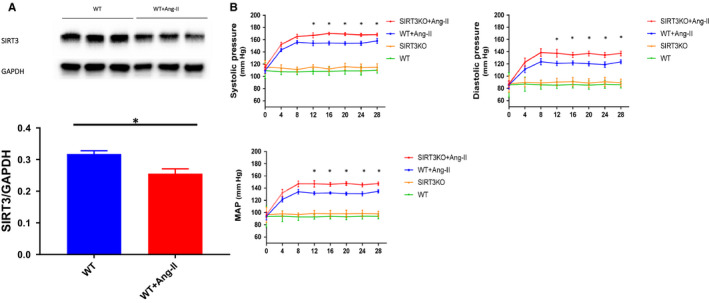
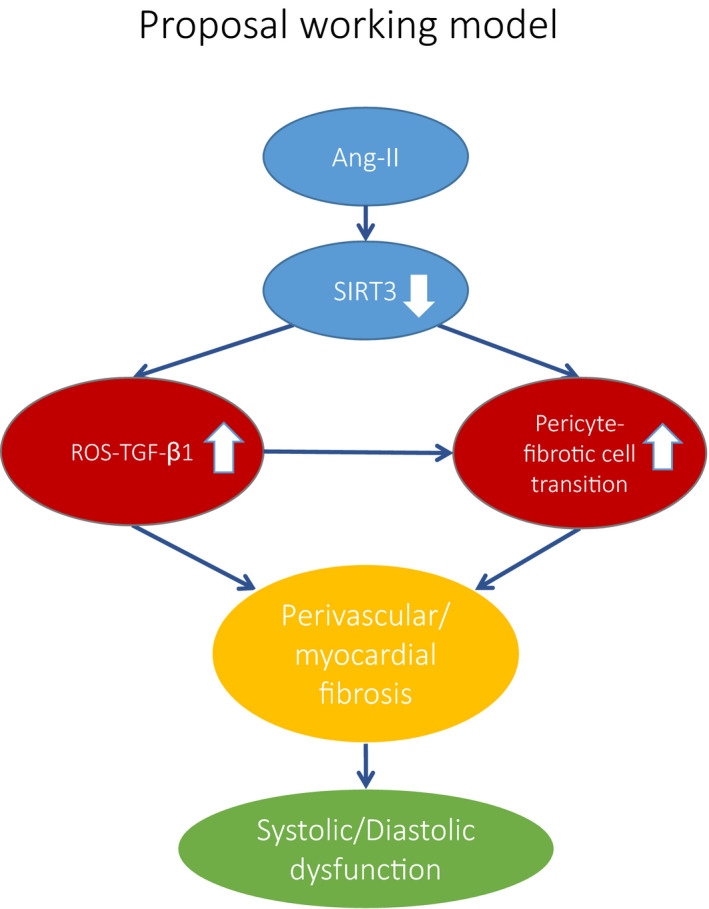
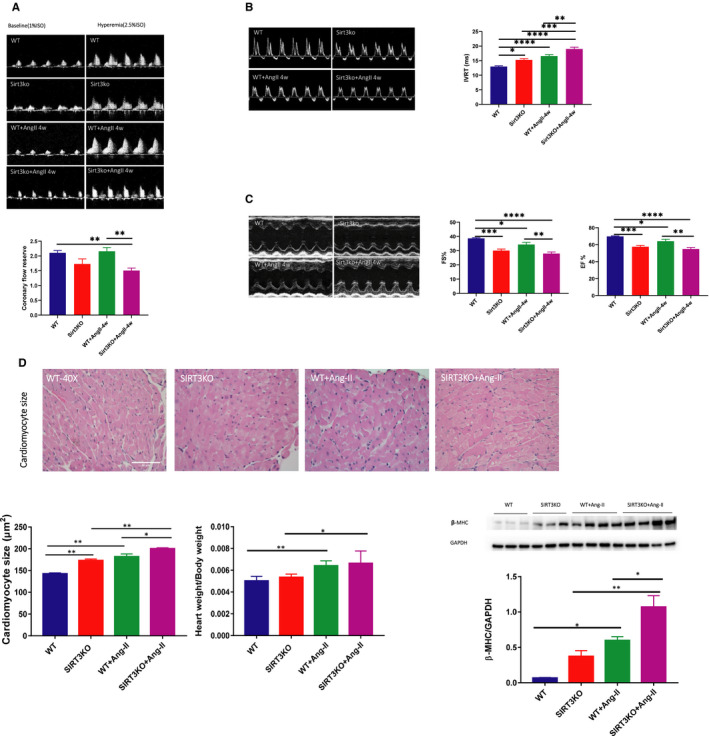
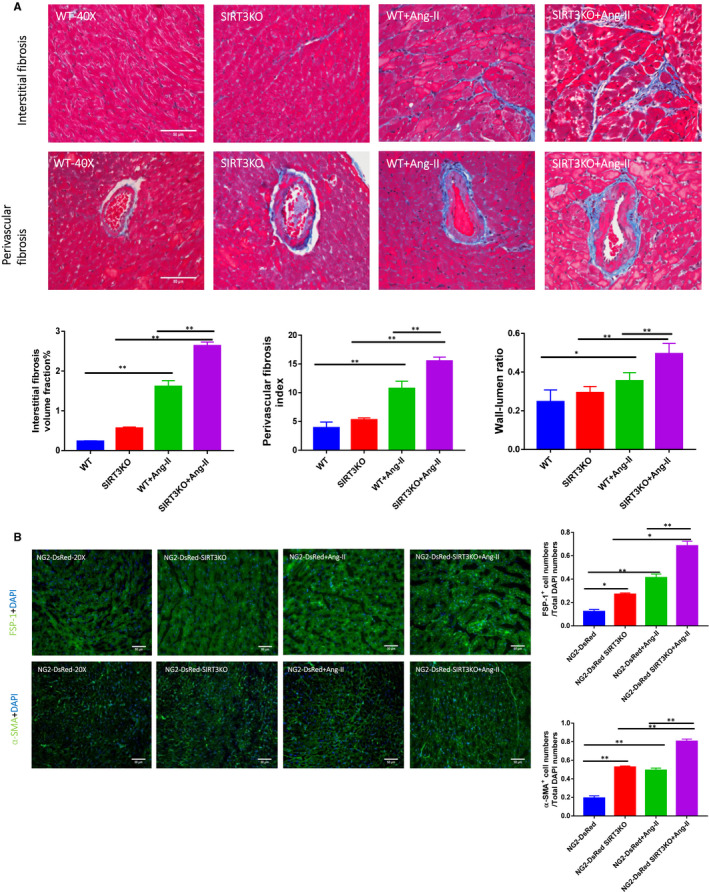
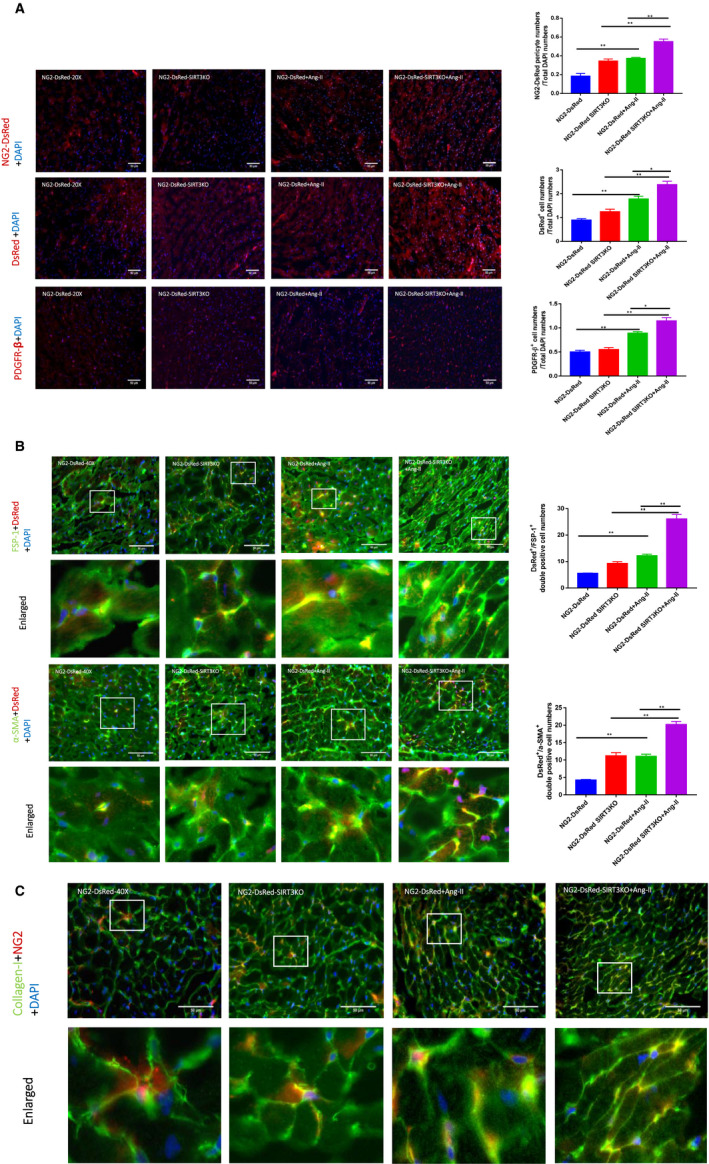
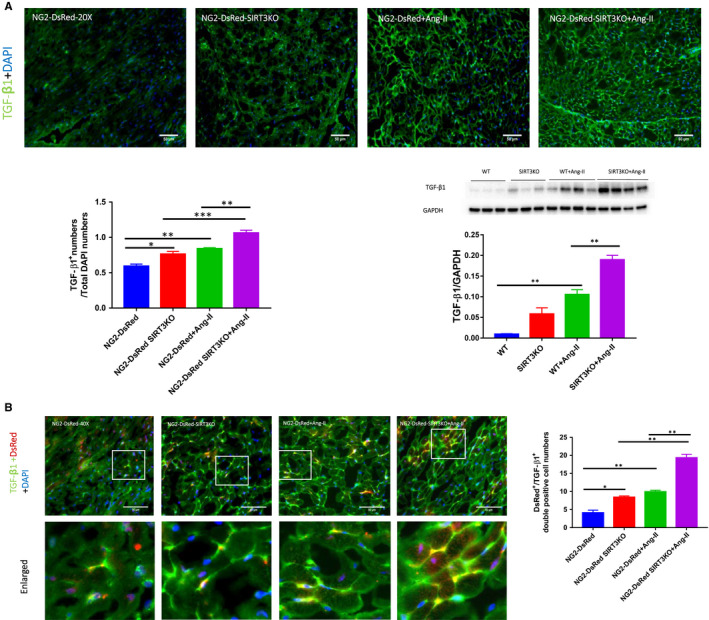
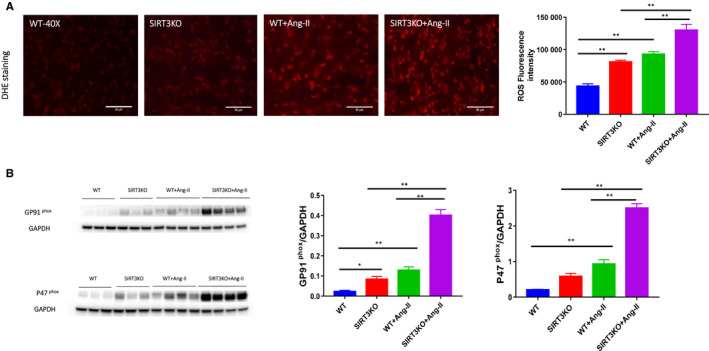
Hypertension is the key factor for the development of cardiac fibrosis and diastolic dysfunction. Our previous study showed that knockout of sirtuin 3 (SIRT3) resulted in diastolic dysfunction in mice. In the present study, we explored the role of SIRT3 in angiotensin II (Ang-II)-induced cardiac fibrosis and pericyte-myofibroblast transition. NG2 tracing reporter NG2-DsRed mouse was crossed with wild-type (WT) mice and SIRT3KO mice. Cardiac function, cardiac fibrosis and reactive oxygen species (ROS) were measured. Mice infused with Ang-II for 28 days showed a significant reduction of SIRT3 expression in the mouse hearts. Knockout of SIRT3 sensitized Ang-II-induced elevation of isovolumic relaxation time (IVRT) and reduction of ejection fraction (EF) and fractional shortening (FS). Ang-II-induced cardiac fibrosis, capillary rarefaction and hypertrophy were further enhanced by knockout of SIRT3. NG2 pericyte tracing reporter mice infused with Ang-II had a significantly increased number of NG2-DsRed pericyte in the heart. Knockout of SIRT3 further enhanced Ang-II-induced increase of pericytes. To examine pericyte-myofibroblast/fibroblast transition, DsRed pericytes were co-stained with FSP-1 and α-SMA. Ang-II infusion led to a significant increase in numbers of DsRed /FSP-1 and DsRed /α-SMA cells, while SIRT3KO further developed pericyte-myofibroblast/fibroblast transition. In addition, knockout of SIRT3 promoted Ang-II-induced NADPH oxidase-derived ROS formation together with increased expression of transforming growth factor beta 1 (TGF-β1). We concluded that Ang-II induced cardiac fibrosis partly by the mechanisms involving SIRT3-mediated pericyte-myofibroblast/fibroblast transition and ROS-TGF-β1 pathway.
高血压是心脏纤维化和舒张功能障碍发展的关键因素。我们之前的研究表明,沉默信息调节因子 3(SIRT3)的敲除会导致小鼠舒张功能障碍。在本研究中,我们探讨了 SIRT3 在血管紧张素 II(Ang-II)诱导的心脏纤维化和周细胞-肌成纤维细胞转化中的作用。NG2 示踪报告基因 NG2-DsRed 小鼠与野生型(WT)和 SIRT3KO 小鼠杂交。测量心脏功能、心脏纤维化和活性氧(ROS)。用 Ang-II 灌注 28 天的小鼠心脏中 SIRT3 的表达明显降低。SIRT3 的敲除使 Ang-II 诱导的等容舒张时间(IVRT)升高和射血分数(EF)和缩短分数(FS)降低更加敏感。SIRT3 的敲除进一步增强了 Ang-II 诱导的心脏纤维化、毛细血管稀疏和肥大。用 Ang-II 灌注的 NG2 周细胞示踪报告基因小鼠心脏中 NG2-DsRed 周细胞数量显著增加。SIRT3 的敲除进一步增强了 Ang-II 诱导的周细胞增加。为了检查周细胞-肌成纤维细胞/成纤维细胞转化,DsRed 周细胞与 FSP-1 和 α-SMA 共染色。Ang-II 灌注导致 DsRed/FSP-1 和 DsRed/α-SMA 细胞数量显著增加,而 SIRT3KO 进一步发展了周细胞-肌成纤维细胞/成纤维细胞转化。此外,SIRT3 的敲除促进了 Ang-II 诱导的 NADPH 氧化酶衍生的 ROS 形成,同时增加了转化生长因子 β1(TGF-β1)的表达。我们得出结论,Ang-II 通过涉及 SIRT3 介导的周细胞-肌成纤维细胞/成纤维细胞转化和 ROS-TGF-β1 途径的机制诱导心脏纤维化。