State Key Laboratory of Medical Genomics, Shanghai Key Laboratory of Hypertension, Department of Hypertension Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China
State Key Laboratory of Medical Genomics, Shanghai Key Laboratory of Hypertension, Department of Hypertension Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
J Am Heart Assoc. 2017 Aug 19;6(8):e006114. doi: 10.1161/JAHA.117.006114.
Emerging evidence indicates that impaired angiogenesis may contribute to hypertension-induced cardiac remodeling. The nicotinamide adenine dinucleotide-dependent deacetylase Sirtuin 3 (SIRT3) has the potential to modulate angiogenesis, but this has not been confirmed. As such, the aim of this study was to examine the relationship between SIRT3-mediated angiogenesis and cardiac remodeling.
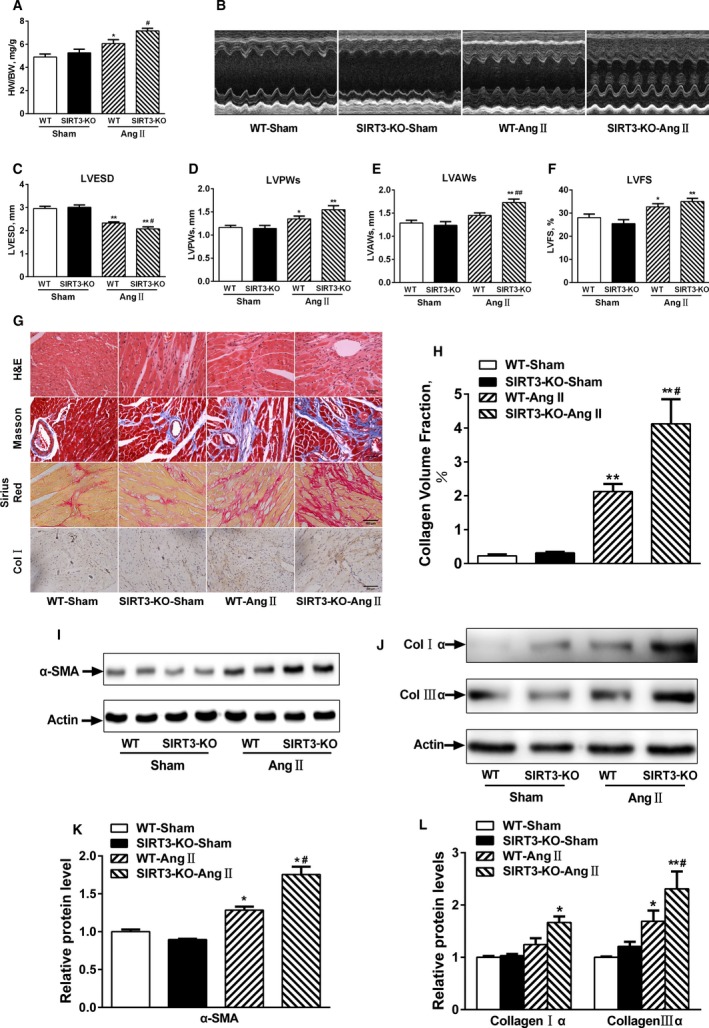
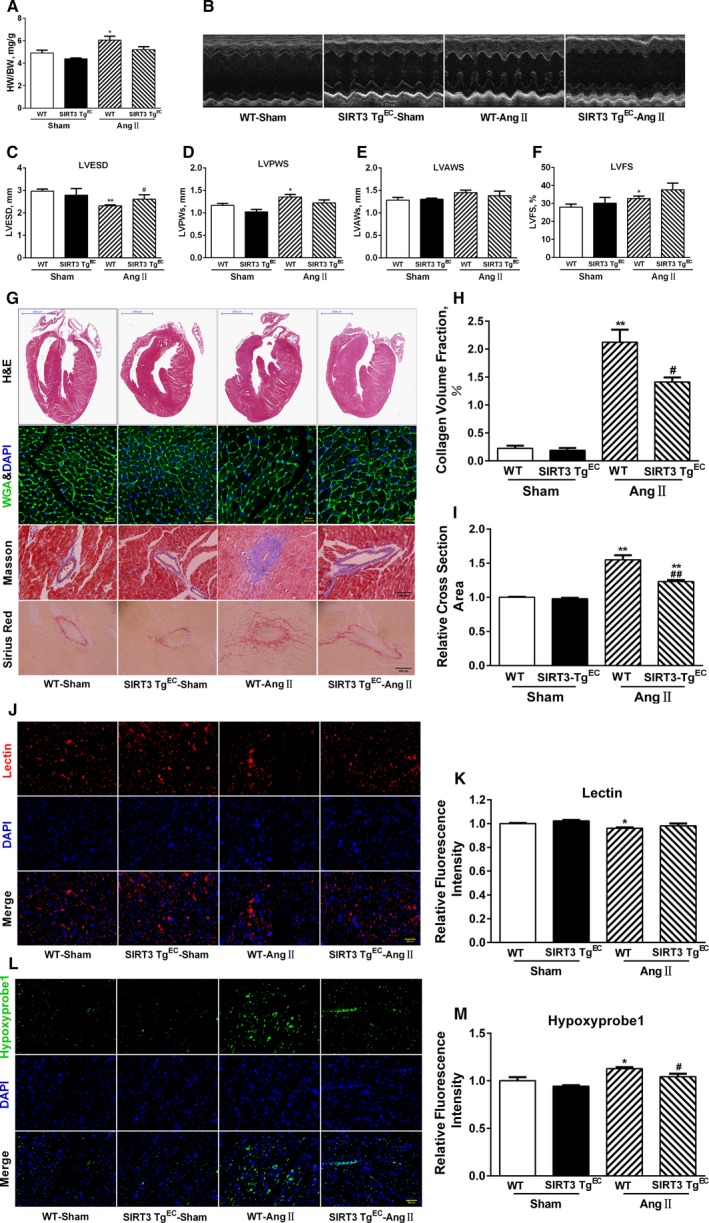
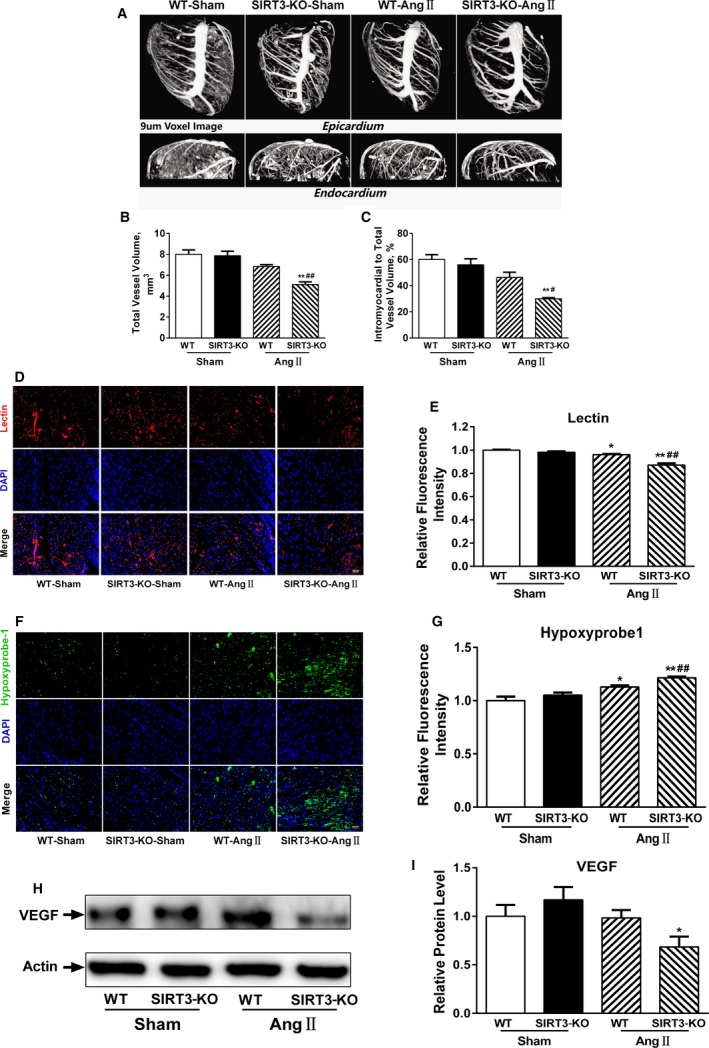
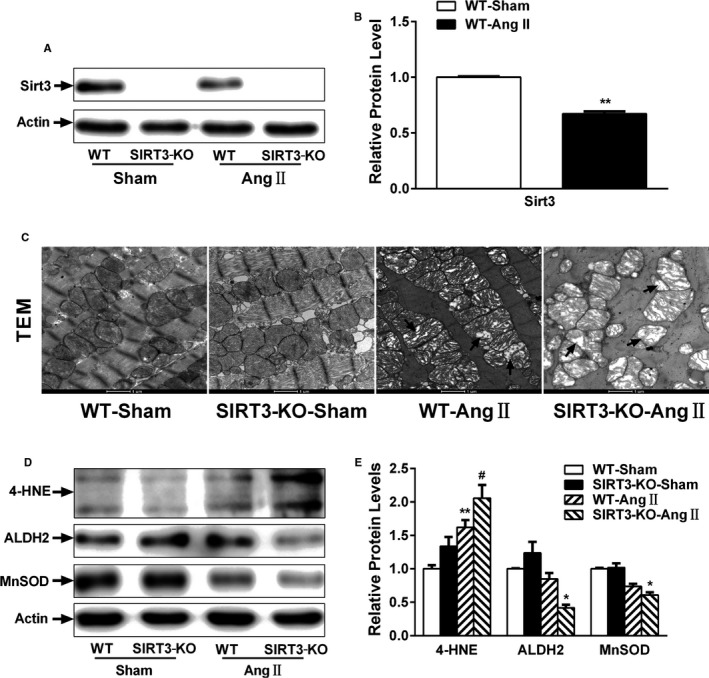
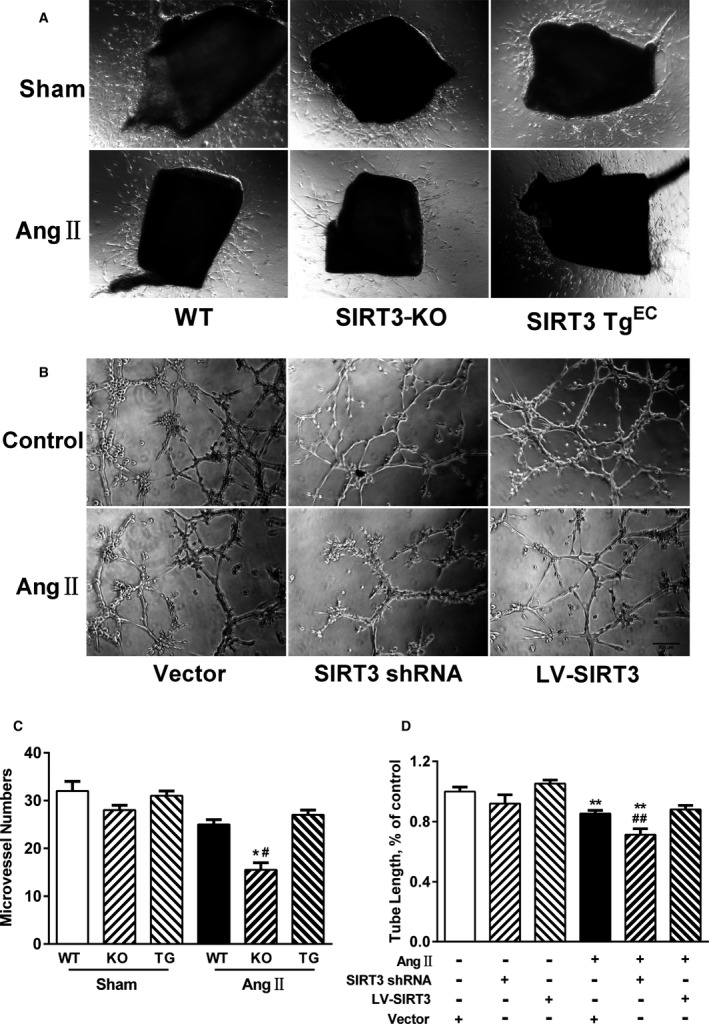
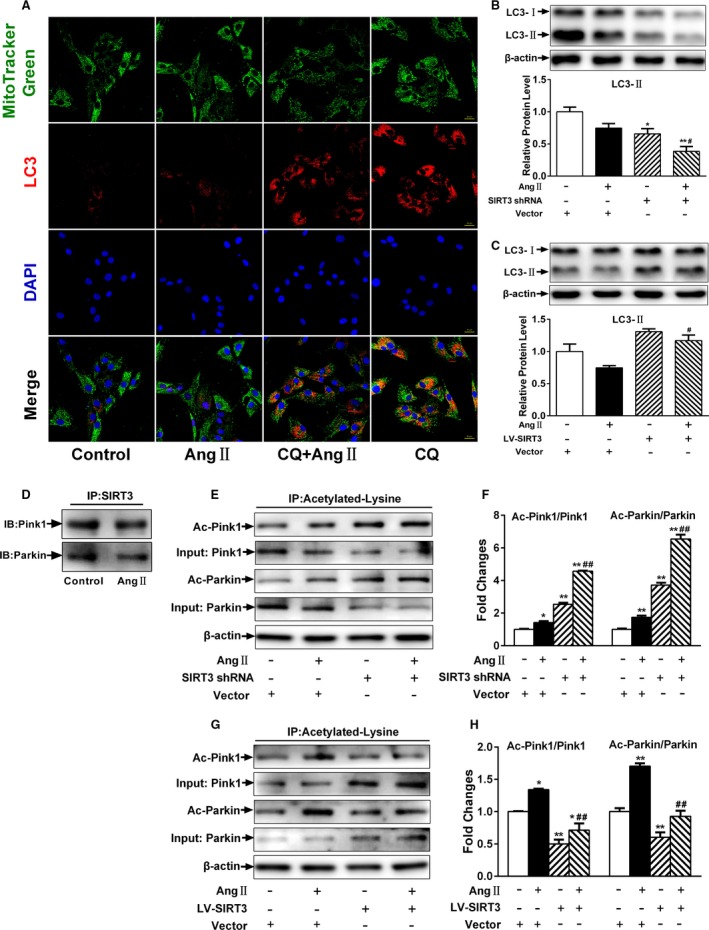
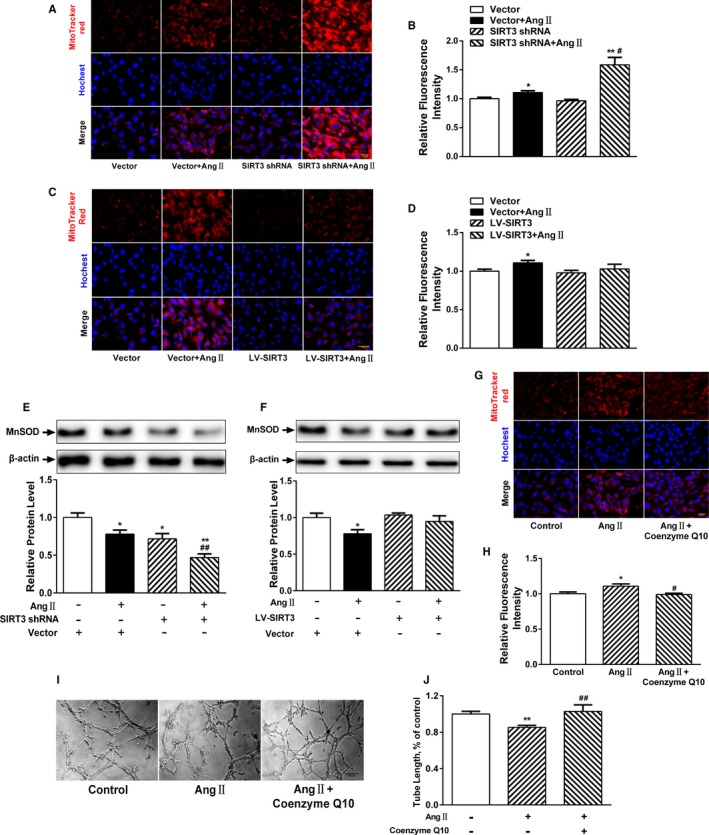
Our experiments were performed on SIRT3 knockout and age-matched wild-type mice infused with angiotensin II (1400 ng/kg per minute) or saline for 14 days. After angiotensin II infusion, SIRT3 knockout mice developed more severe microvascular rarefaction and functional hypoxia in cardiac tissues compared with wild-type mice. These events were concomitant with mitochondrial dysfunction and enhanced collagen I and collagen III expression, leading to cardiac fibrosis. Silencing SIRT3 facilitated angiotensin II-induced aberrant Pink/Parkin acetylation and impaired mitophagy, while excessive mitochondrial reactive oxygen species generation limited angiogenic capacity in primary mouse cardiac microvascular endothelial cells. Moreover, SIRT3 overexpression in cardiac microvascular endothelial cells enhanced Pink/Parkin-mediated mitophagy, attenuated mitochondrial reactive oxygen species generation, and restored vessel sprouting and tube formation. In parallel, endothelial cell-specific SIRT3 transgenic mice showed decreased fibrosis, as well as improved cardiac function and microvascular network, compared with wild-type mice with similar stimuli.
Collectively, these findings suggest that SIRT3 could promote angiogenesis through attenuating mitochondrial dysfunction caused by defective mitophagy.
新出现的证据表明,血管生成受损可能导致高血压引起的心脏重构。烟酰胺腺嘌呤二核苷酸依赖性去乙酰化酶 Sirtuin 3(SIRT3)具有调节血管生成的潜力,但这尚未得到证实。因此,本研究旨在研究 SIRT3 介导的血管生成与心脏重构之间的关系。
我们在 SIRT3 敲除和年龄匹配的野生型小鼠中进行了实验,这些小鼠接受血管紧张素 II(1400ng/kg/分钟)或生理盐水输注 14 天。血管紧张素 II 输注后,SIRT3 敲除小鼠的心脏组织中微血管稀疏和功能缺氧比野生型小鼠更为严重。这些事件伴随着线粒体功能障碍和胶原 I 和胶原 III 表达增强,导致心脏纤维化。沉默 SIRT3 促进了血管紧张素 II 诱导的 Pink/Parkin 乙酰化和受损的线粒体自噬,而过量的线粒体活性氧生成限制了原代小鼠心脏微血管内皮细胞的血管生成能力。此外,心脏微血管内皮细胞中 SIRT3 的过表达增强了 Pink/Parkin 介导的线粒体自噬,减轻了线粒体活性氧的产生,并恢复了血管发芽和管形成。此外,与接受类似刺激的野生型小鼠相比,内皮细胞特异性 SIRT3 转基因小鼠的纤维化减少,心脏功能和微血管网络得到改善。
综上所述,这些发现表明 SIRT3 可以通过减轻缺陷性线粒体自噬引起的线粒体功能障碍来促进血管生成。