APHP, Service de Neuropédiatrie, Hôpital Armand Trousseau, Sorbonne Université, 75012, Paris, France.
Centre de Référence Neurogénétique, Mouvement Anormaux de l'enfant, Hopital Armand Trousseau, 75012, Paris, France.
Eur J Hum Genet. 2020 Oct;28(10):1403-1413. doi: 10.1038/s41431-020-0641-9. Epub 2020 May 28.
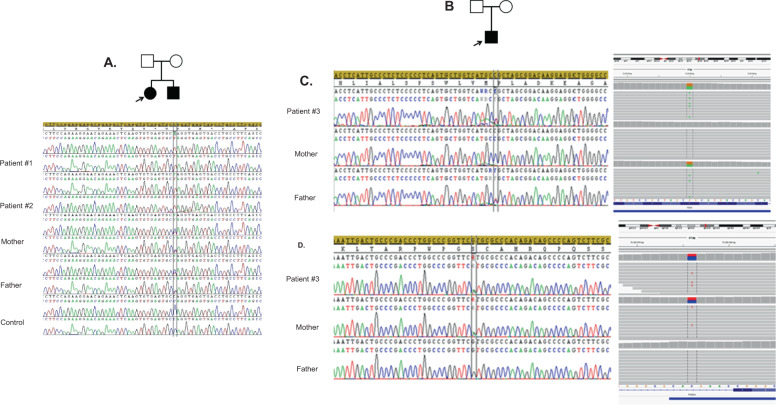
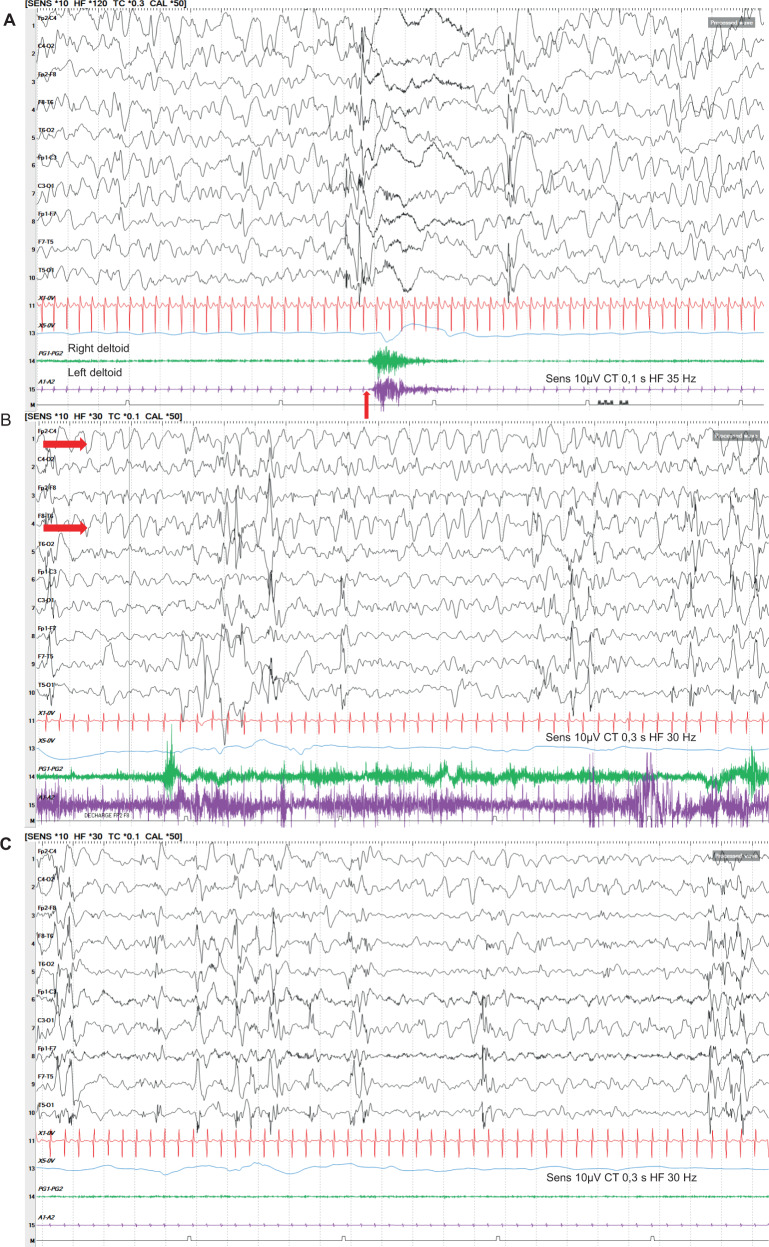
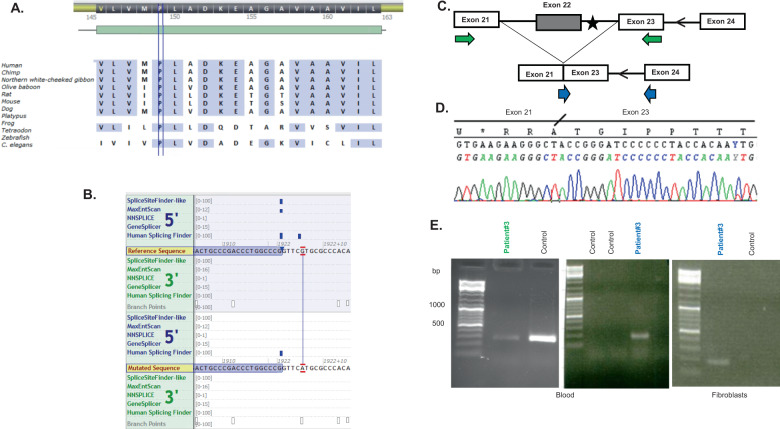
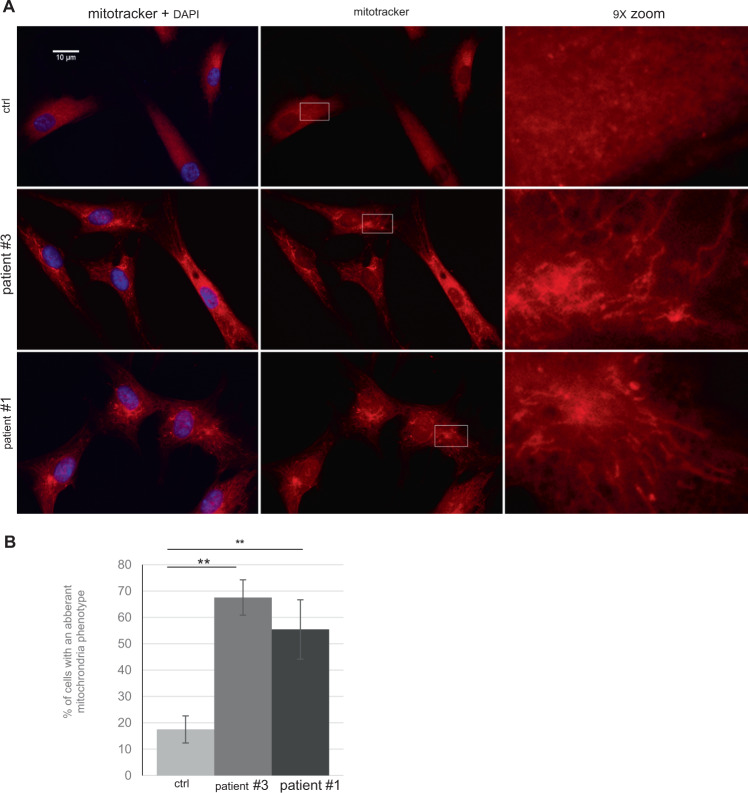
Cause of complex dyskinesia remains elusive in some patients. A homozygous missense variant leading to drastic decrease of PDE2A enzymatic activity was reported in one patient with childhood-onset choreodystonia preceded by paroxysmal dyskinesia and associated with cognitive impairment and interictal EEG abnormalities. Here, we report three new cases with biallelic PDE2A variants identified by trio whole-exome sequencing. Mitochondria network was analyzed after Mitotracker™ Red staining in control and mutated primary fibroblasts. Analysis of retrospective video of patients' movement disorder and refinement of phenotype was carried out. We identified a homozygous gain of stop codon variant c.1180C>T; p.(Gln394*) in PDE2A in siblings and compound heterozygous variants in young adult: a missense c.446C>T; p.(Pro149Leu) and splice-site variant c.1922+5G>A predicted and shown to produce an out of frame transcript lacking exon 22. All three patients had cognitive impairment or developmental delay. The phenotype of the two oldest patients, aged 9 and 26, was characterized by childhood-onset refractory paroxysmal dyskinesia initially misdiagnosed as epilepsy due to interictal EEG abnormalities. The youngest patient showed a proven epilepsy at the age of 4 months and no paroxysmal dyskinesia at 15 months. Interestingly, analysis of the fibroblasts with the biallelic variants in PDE2A variants revealed mitochondria network morphology changes. Together with previously reported case, our three patients confirm that biallelic PDE2A variants are a cause of childhood-onset refractory paroxysmal dyskinesia with cognitive impairment, sometimes associated with choreodystonia and interictal baseline EEG abnormalities or epilepsy.
在一些患者中,复杂运动障碍的病因仍然难以捉摸。曾有报道称,一名儿童期起病的舞蹈徐动症患者伴有阵发性运动障碍,伴有认知障碍和发作间期脑电图异常,存在导致 PDE2A 酶活性急剧下降的纯合错义变异。在此,我们报告了通过 trio 全外显子组测序鉴定的 3 例新的 PDE2A 双等位基因变异病例。在对照和突变的原代成纤维细胞中用 Mitotracker™ Red 染色后分析线粒体网络。对患者运动障碍的回顾性视频进行分析并对表型进行细化。我们在兄弟姐妹中鉴定出 PDE2A 的纯合终止密码子变异 c.1180C>T;p.(Gln394*),在年轻成人中鉴定出复合杂合变异:错义 c.446C>T;p.(Pro149Leu)和剪接位点变异 c.1922+5G>A,预测并显示产生缺少外显子 22 的无框转录本。所有三名患者均有认知障碍或发育迟缓。两名年龄最大的患者(9 岁和 26 岁)的表型特征为儿童期起病的难治性阵发性运动障碍,最初由于发作间期脑电图异常被误诊为癫痫。最小的患者在 4 个月时表现为已确诊的癫痫,15 个月时无阵发性运动障碍。有趣的是,对具有双等位基因 PDE2A 变异的成纤维细胞进行分析显示线粒体网络形态发生变化。结合以前报道的病例,我们的 3 例患者证实,双等位基因 PDE2A 变异是儿童期起病的难治性阵发性运动障碍伴认知障碍的原因,有时伴有舞蹈徐动症和发作间期基线脑电图异常或癫痫。