School of Biological Sciences, University of the Punjab Quaid-i-Azam Campus, Lahore, Pakistan.
Medical Genetics, Mugla Sitki Kocman University Faculty of Medicine, Mugla, Turkey.
J Med Genet. 2021 Apr;58(4):237-246. doi: 10.1136/jmedgenet-2020-106849. Epub 2020 May 21.
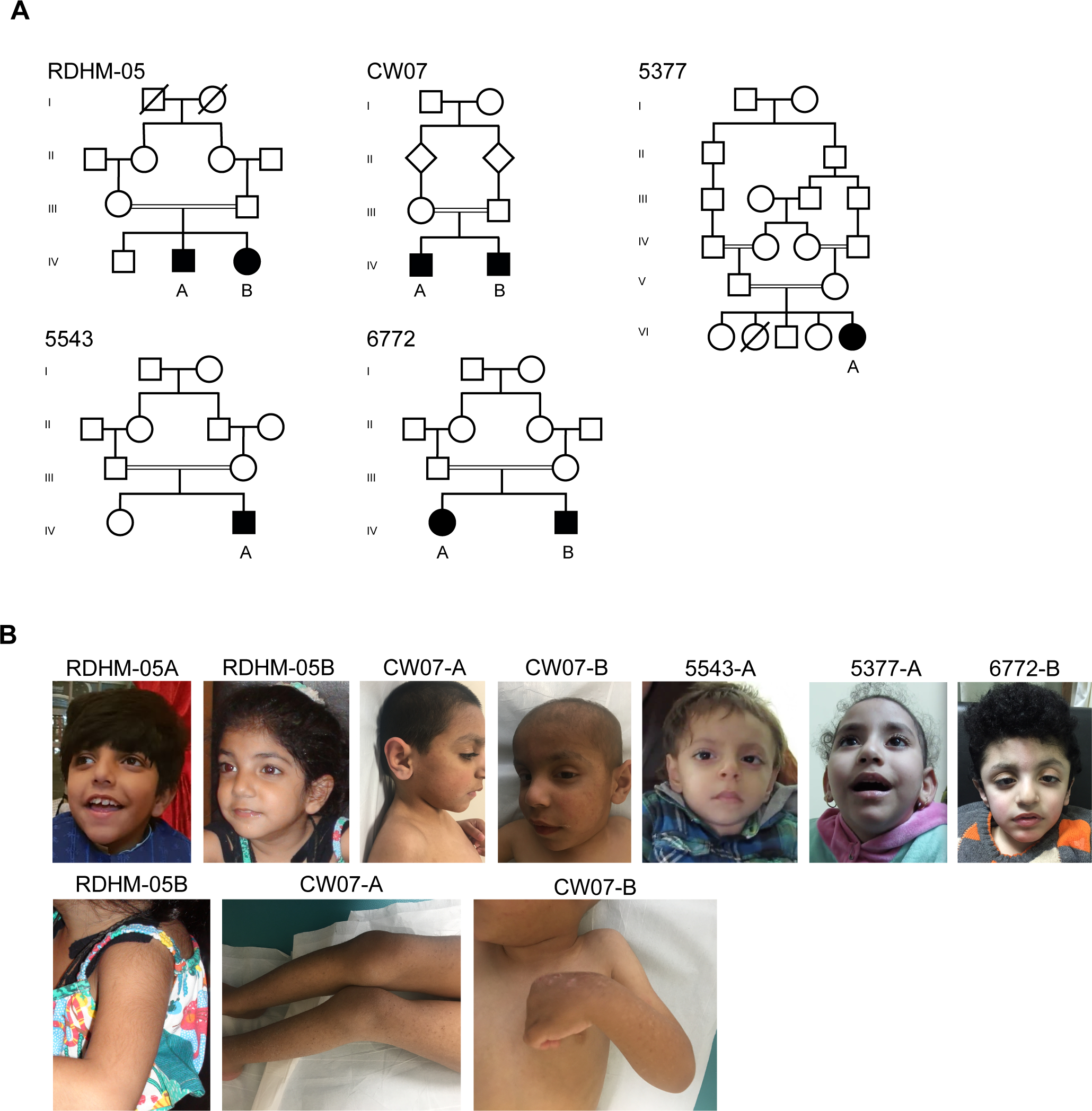
Intellectual disability syndromes (IDSs) with or without developmental delays affect up to 3% of the world population. We sought to clinically and genetically characterise a novel IDS segregating in five unrelated consanguineous families.
Clinical analyses were performed for eight patients with intellectual disability (ID). Whole-exome sequencing for selected participants followed by Sanger sequencing for all available family members was completed. Identity-by-descent (IBD) mapping was carried out for patients in two Egyptian families harbouring an identical variant. RNA was extracted from blood cells of Turkish participants, followed by cDNA synthesis and real-time PCR for .
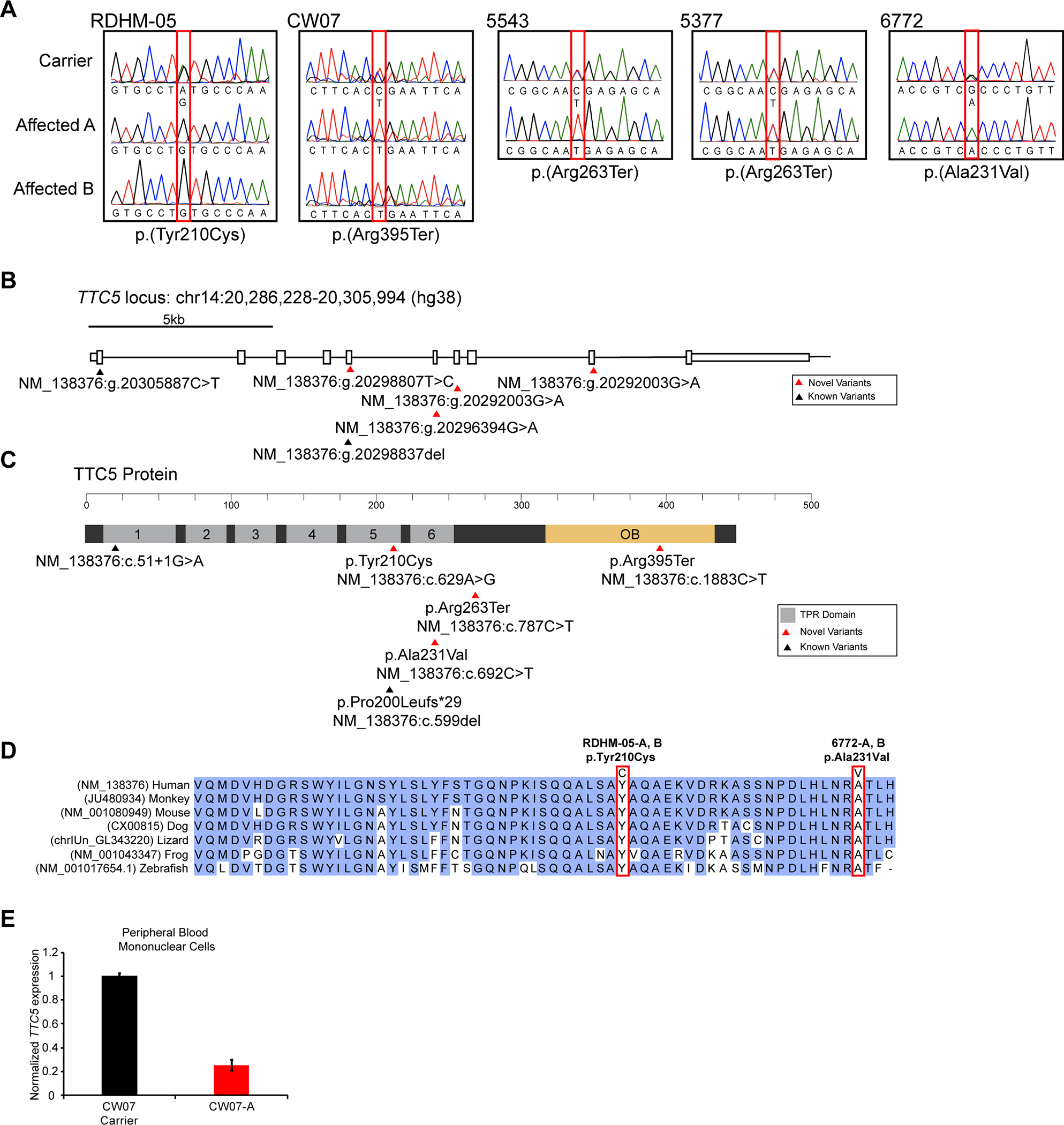
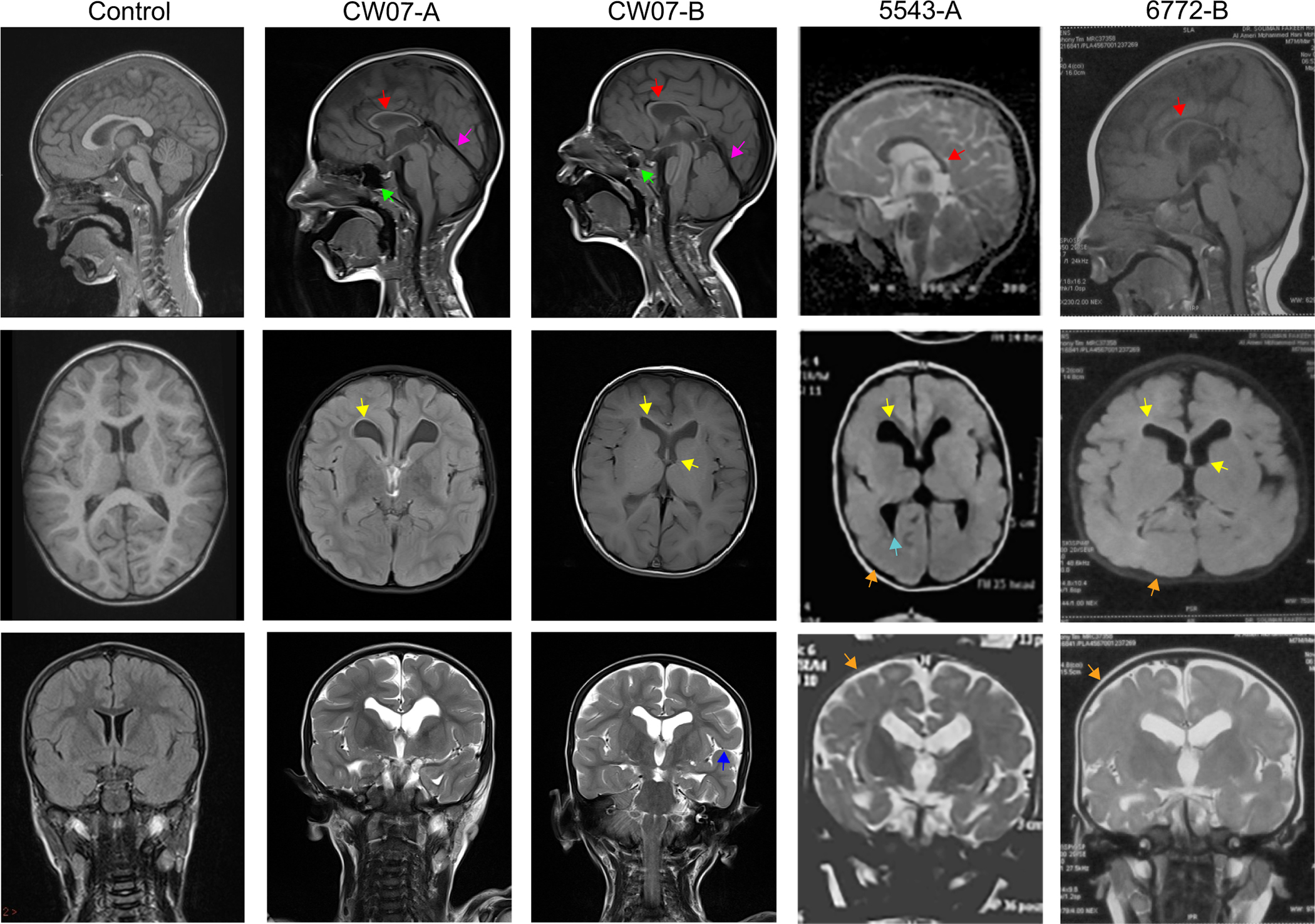
Phenotype comparisons of patients revealed shared clinical features of moderate-to-severe ID, corpus callosum agenesis, mild ventriculomegaly, simplified gyral pattern, cerebral atrophy, delayed motor and verbal milestones and hypotonia, presenting with an IDS. Four novel homozygous variants in : c.629A>G;p.(Tyr210Cys), c.692C>T;p.(Ala231Val), c.787C>T;p.(Arg263Ter) and c.1883C>T;p.(Arg395Ter) were identified in the eight patients from participating families. IBD mapping revealed that c.787C>T;p.(Arg263Ter) is a founder variant in Egypt. Missense variants c.629A>G;p.(Tyr210Cys) and c.692C>T;p.(Ala231Val) disrupt highly conserved residues of TTC5 within the fifth and sixth tetratricopeptide repeat motifs which are required for p300 interaction, while the nonsense variants are predicted to decrease expression. Functional analysis of variant c.1883C>T;p.(Arg395Ter) showed reduced transcript levels in accordance with nonsense-mediated decay.
Combining our clinical and molecular data with a recent case report, we identify the core and variable clinical features associated with loss-of-function variants and reveal the requirement for TTC5 in human brain development and health.
伴有或不伴有发育迟缓的智力障碍综合征(IDSs)影响了全球多达 3%的人口。我们试图对五个无关近亲家庭中分离的新型 IDS 进行临床和基因特征分析。
对 8 名智力障碍(ID)患者进行临床分析。对选定的参与者进行全外显子组测序,然后对所有可用的家族成员进行 Sanger 测序。对两个埃及家庭的患者进行同源性区域(IBD)映射,这两个家庭存在相同的变异。从土耳其参与者的血细胞中提取 RNA,然后进行 cDNA 合成和实时 PCR 检测 。
对患者的表型比较显示,共享中度至重度 ID、胼胝体发育不全、轻度脑室扩大、简化脑回模式、脑萎缩、运动和语言发育迟缓以及张力减退的共同临床特征,表现为 IDS。在参与家庭的 8 名患者中发现了 4 个新的纯合变异:c.629A>G;p.(Tyr210Cys)、c.692C>T;p.(Ala231Val)、c.787C>T;p.(Arg263Ter)和 c.1883C>T;p.(Arg395Ter)。IBD 映射显示 c.787C>T;p.(Arg263Ter)是埃及的一个创始变异。错义变异 c.629A>G;p.(Tyr210Cys)和 c.692C>T;p.(Ala231Val)破坏了第五和第六四肽重复基序中高度保守的 TTC5 残基,这对于 p300 相互作用是必需的,而无义变异则预计会降低 表达。对变异 c.1883C>T;p.(Arg395Ter)的功能分析显示,与无义介导的衰变一致, 转录物水平降低。
结合我们的临床和分子数据以及最近的一个病例报告,我们确定了与 TTC5 功能丧失变异相关的核心和可变临床特征,并揭示了 TTC5 在人类大脑发育和健康中的必要性。