IBIMA, Rostock University Medical Center, Rostock, 18041, Germany.
SHIP-KEF, Institute for Community Medicine, University Medicine of Greifswald, Walther-Rathenau-Straβe 48, 17475 Greifswald, Germany.
Brief Bioinform. 2021 May 20;22(3). doi: 10.1093/bib/bbaa072.
The difficulty to find new drugs and bring them to the market has led to an increased interest to find new applications for known compounds. Biological samples from many disease contexts have been extensively profiled by transcriptomics, and, intuitively, this motivates to search for compounds with a reversing effect on the expression of characteristic disease genes. However, disease effects may be cell line-specific and also depend on other factors, such as genetics and environment. Transcription profile changes between healthy and diseased cells relate in complex ways to profile changes gathered from cell lines upon stimulation with a drug. Despite these differences, we expect that there will be some similarity in the gene regulatory networks at play in both situations. The challenge is to match transcriptomes for both diseases and drugs alike, even though the exact molecular pathology/pharmacogenomics may not be known.
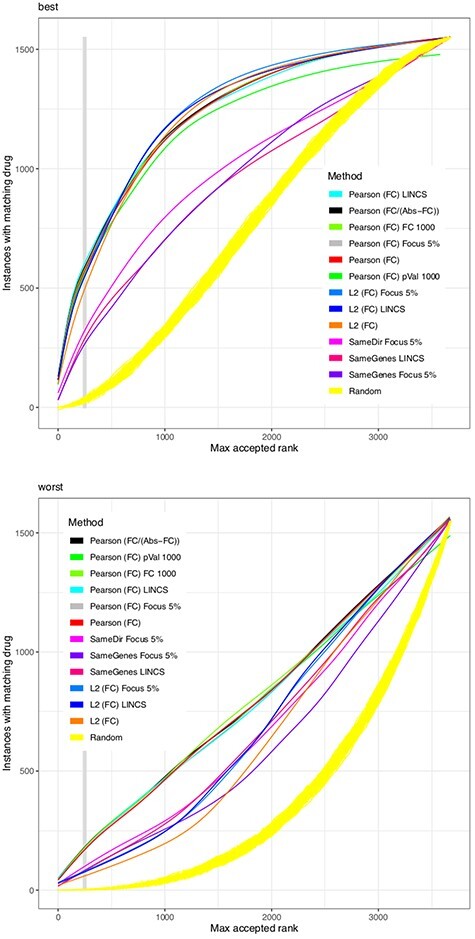
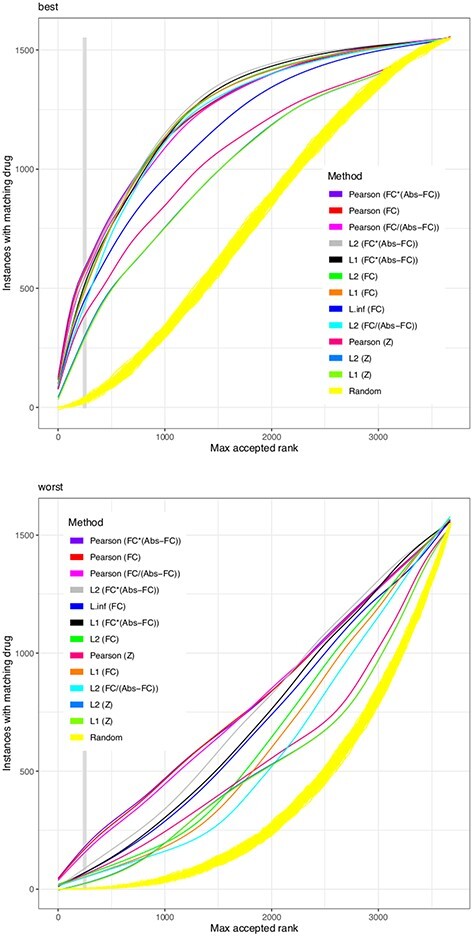
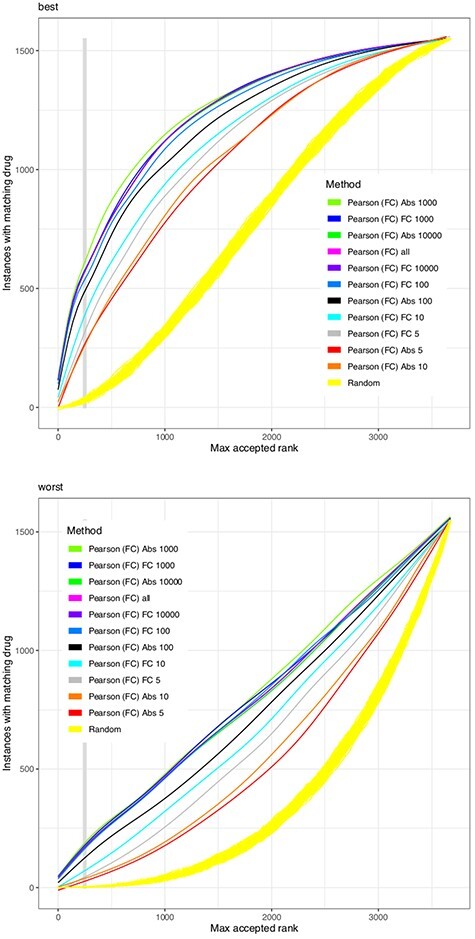
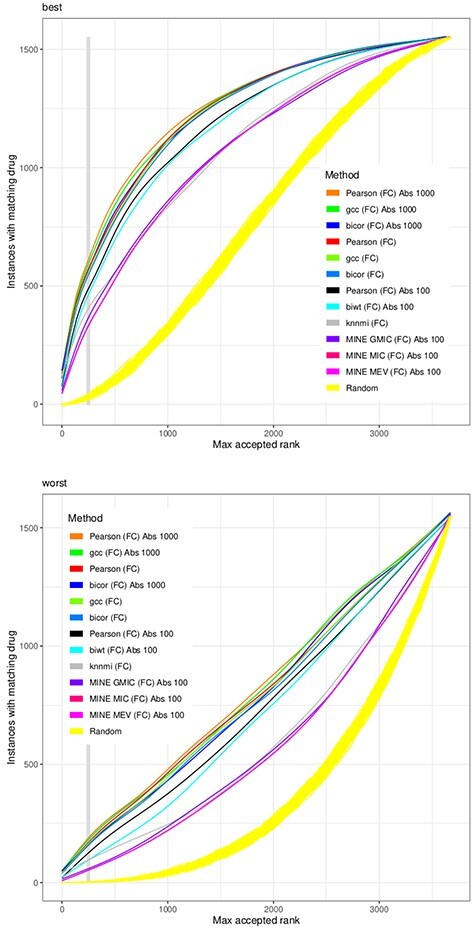
We substitute the challenge to match a drug effect to a disease effect with the challenge to match a drug effect to the effect of the same drug at another concentration or in another cell line. This is welldefined, reproducible in vitro and in silico and extendable with external data. Based on the Connectivity Map (CMap) dataset, we combined 26 different similarity scores with six different heuristics to reduce the number of genes in the model. Such gene filters may also utilize external knowledge e.g. from biological networks. We found that no similarity score always outperforms all others for all drugs, but the Pearson correlation finds the same drug with the highest reliability. Results are improved by filtering for highly expressed genes and to a lesser degree for genes with large fold changes. Also a network-based reduction of contributing transcripts was beneficial, here implemented by the FocusHeuristics. We found no drop in prediction accuracy when reducing the whole transcriptome to the set of 1000 landmark genes of the CMap's successor project Library of Integrated Network-based Cellular Signatures. All source code to re-analyze and extend the CMap data, the source code of heuristics, filters and their evaluation are available to propel the development of new methods for drug repurposing.
https://bitbucket.org/ibima/moldrugeffectsdb.
steffen.moeller@uni-rostock.de.
Supplementary data are available at Briefings in Bioinformatics online.
寻找新药并将其推向市场的困难导致人们越来越关注为已知化合物寻找新的应用。通过转录组学对来自许多疾病背景的生物样本进行了广泛的分析,直观地说,这促使人们寻找对特征疾病基因表达具有逆转作用的化合物。然而,疾病的影响可能是细胞系特异性的,也可能取决于其他因素,如遗传和环境。健康细胞和患病细胞之间的转录谱变化与药物刺激后从细胞系中收集的谱变化以复杂的方式相关。尽管存在这些差异,但我们期望在两种情况下起作用的基因调控网络会有一些相似之处。挑战在于匹配疾病和药物的转录组,即使确切的分子病理学/药物基因组学尚不清楚。
我们将匹配药物作用与疾病作用的挑战替换为匹配药物作用与同一药物在另一种浓度或另一种细胞系中的作用的挑战。这在体外和计算机上定义明确,可重复且可通过外部数据扩展。基于 Connectivity Map(CMap)数据集,我们结合了 26 种不同的相似度得分和 6 种不同的启发式方法来减少模型中的基因数量。此类基因过滤器还可以利用外部知识,例如来自生物网络的知识。我们发现,对于所有药物,没有一种相似度得分总是优于所有其他得分,但 Pearson 相关性以最高的可靠性找到了相同的药物。通过过滤高表达基因和在较小程度上过滤具有较大倍数变化的基因,可以提高结果。基于网络的转录本减少也很有益,这里通过 FocusHeuristics 实现。当将整个转录组减少到 CMap 后继项目 Library of Integrated Network-based Cellular Signatures 的 1000 个标志性基因集时,我们发现预测准确性没有下降。重新分析和扩展 CMap 数据的所有源代码、启发式方法、过滤器及其评估的源代码均可用于推动药物再利用新方法的发展。
https://bitbucket.org/ibima/moldrugeffectsdb.
steffen.moeller@uni-rostock.de.
补充数据可在Briefings in Bioinformatics 在线获取。