Department of Pediatrics, Washington University School of Medicine, St. Louis, Missouri, United States of America.
University of North Carolina-Wilmington, Wilmington, North Carolina, United States of America.
PLoS Pathog. 2020 Jun 4;16(6):e1007806. doi: 10.1371/journal.ppat.1007806. eCollection 2020 Jun.
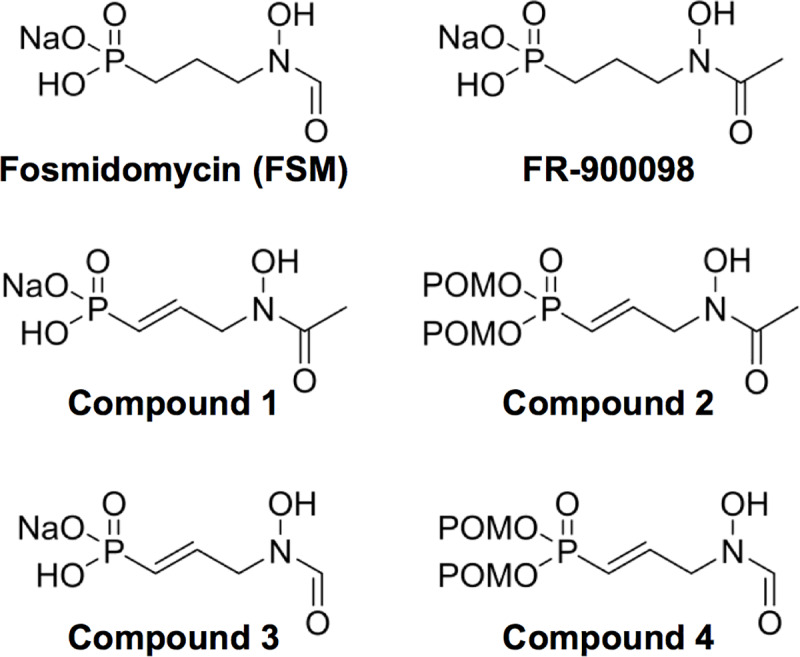
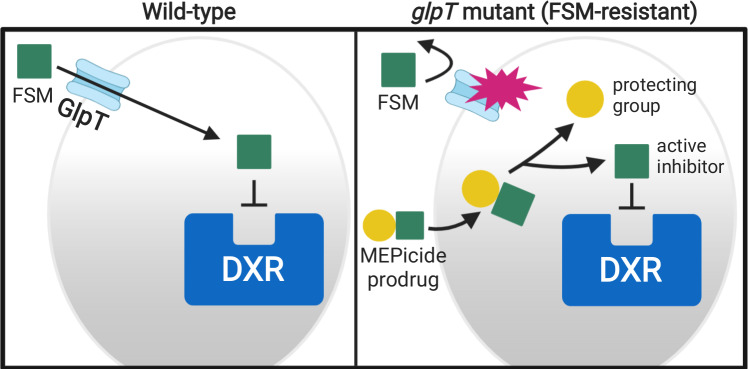
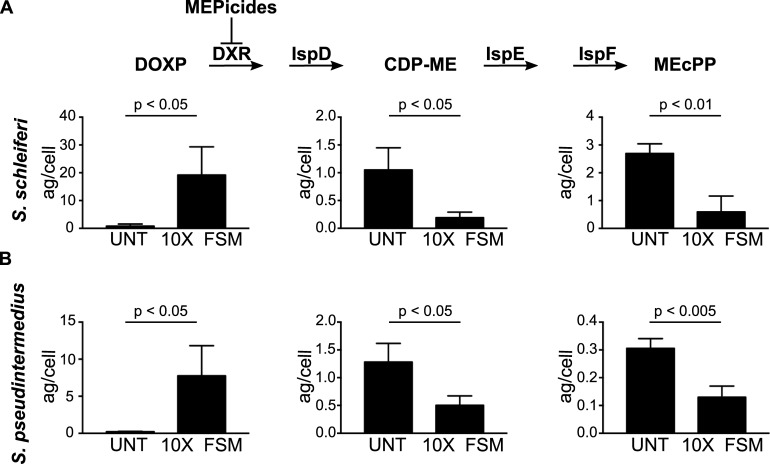
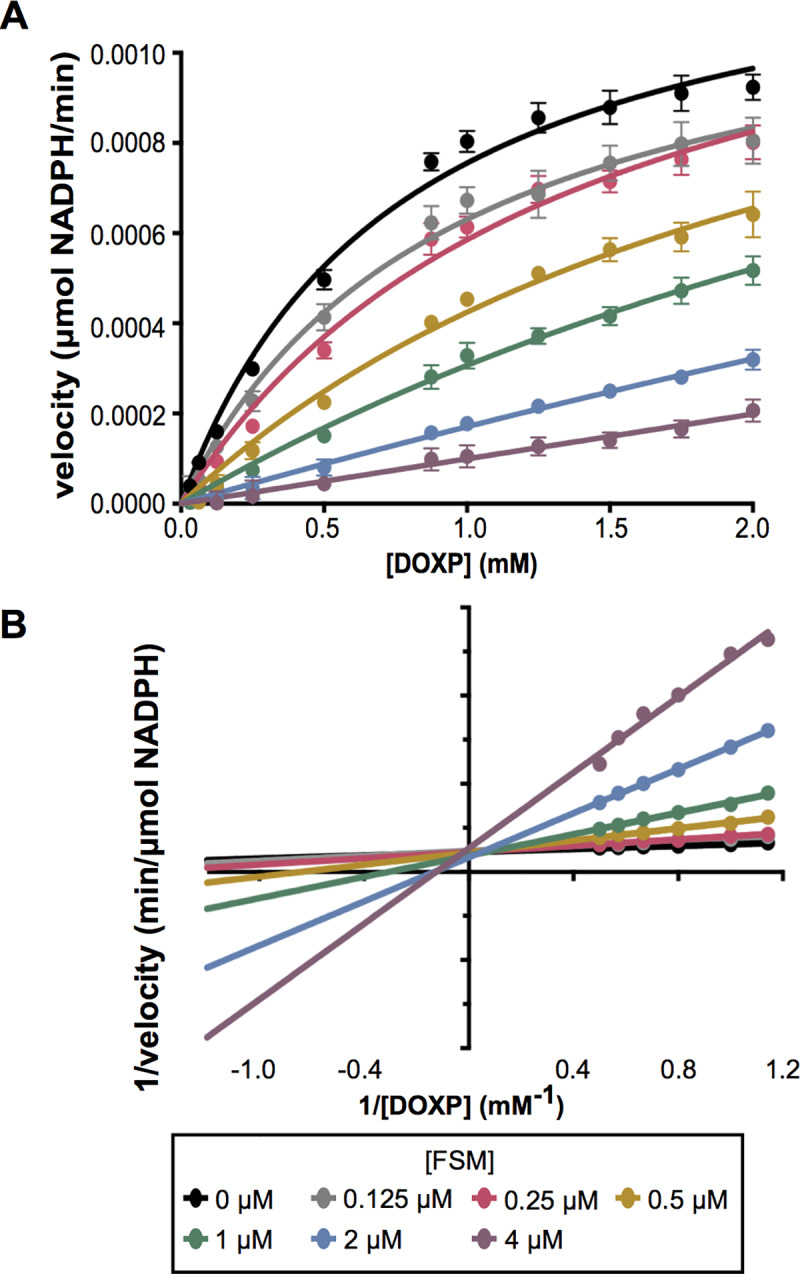
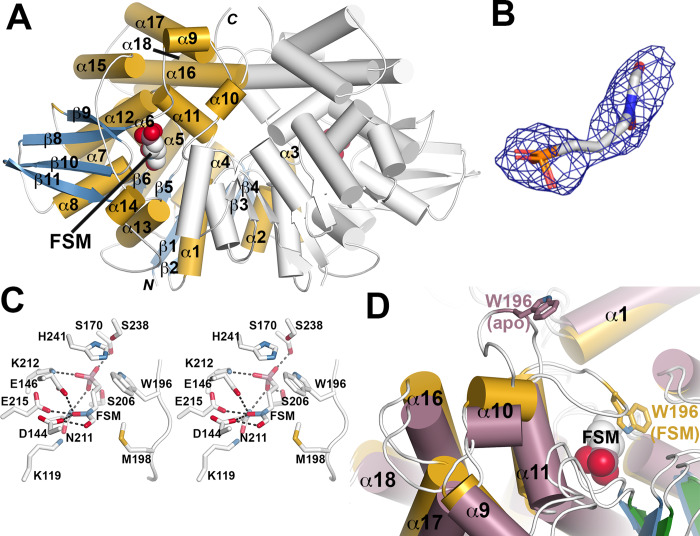
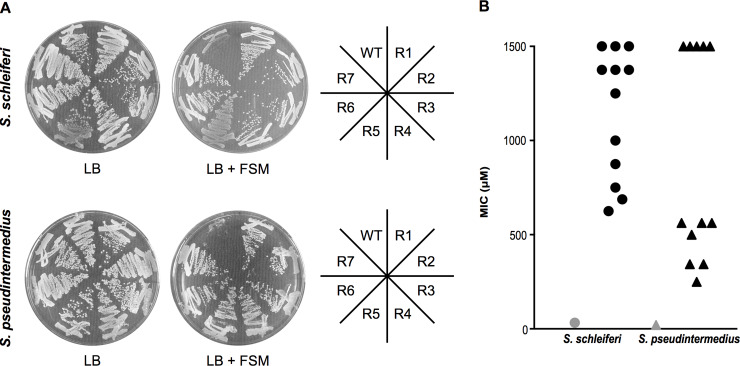
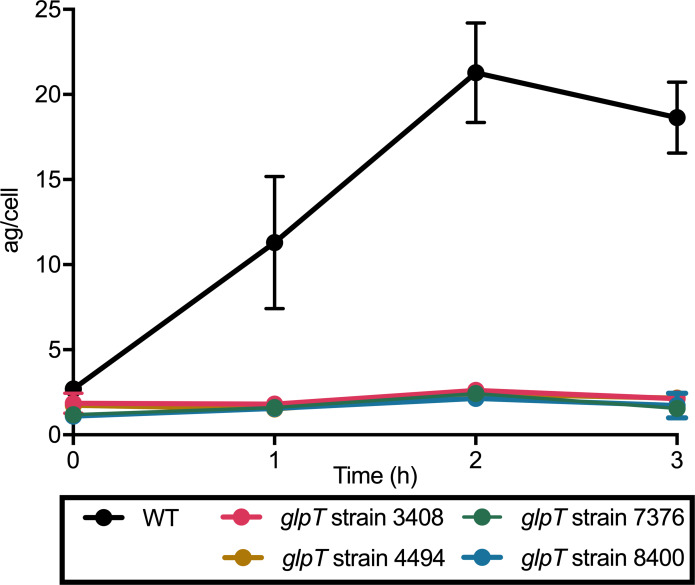
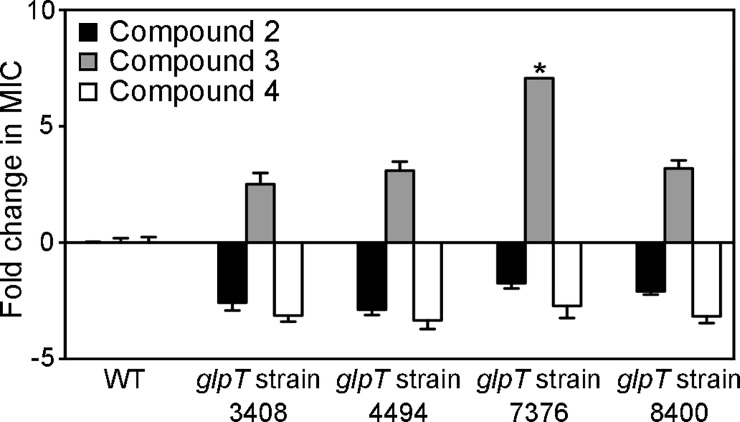
Coagulase-positive staphylococci, which frequently colonize the mucosal surfaces of animals, also cause a spectrum of opportunistic infections including skin and soft tissue infections, urinary tract infections, pneumonia, and bacteremia. However, recent advances in bacterial identification have revealed that these common veterinary pathogens are in fact zoonoses that cause serious infections in human patients. The global spread of multidrug-resistant zoonotic staphylococci, in particular the emergence of methicillin-resistant organisms, is now a serious threat to both animal and human welfare. Accordingly, new therapeutic targets that can be exploited to combat staphylococcal infections are urgently needed. Enzymes of the methylerythritol phosphate pathway (MEP) of isoprenoid biosynthesis represent potential targets for treating zoonotic staphylococci. Here we demonstrate that fosmidomycin (FSM) inhibits the first step of the isoprenoid biosynthetic pathway catalyzed by deoxyxylulose phosphate reductoisomerase (DXR) in staphylococci. In addition, we have both enzymatically and structurally determined the mechanism by which FSM elicits its effect. Using a forward genetic screen, the glycerol-3-phosphate transporter GlpT that facilitates FSM uptake was identified in two zoonotic staphylococci, Staphylococcus schleiferi and Staphylococcus pseudintermedius. A series of lipophilic ester prodrugs (termed MEPicides) structurally related to FSM were synthesized, and data indicate that the presence of the prodrug moiety not only substantially increased potency of the inhibitors against staphylococci but also bypassed the need for GlpT-mediated cellular transport. Collectively, our data indicate that the prodrug MEPicides selectively and robustly inhibit DXR in zoonotic staphylococci, and further, that DXR represents a promising, druggable target for future development.
凝固酶阳性葡萄球菌经常定植于动物的黏膜表面,也会引起一系列机会性感染,包括皮肤和软组织感染、尿路感染、肺炎和菌血症。然而,细菌鉴定的最新进展表明,这些常见的兽医病原体实际上是人畜共患病病原体,会导致人类患者发生严重感染。多药耐药性人畜共患病葡萄球菌的全球传播,特别是耐甲氧西林生物体的出现,现在对动物和人类健康都构成了严重威胁。因此,迫切需要寻找新的治疗靶点来对抗葡萄球菌感染。异戊烯基磷酸途径(MEP)的酶是治疗人畜共患病葡萄球菌的潜在靶点。在这里,我们证明福司霉素(FSM)抑制了葡萄球菌脱氧木酮糖磷酸还原异构酶(DXR)催化的异戊烯基生物合成途径的第一步。此外,我们还从酶学和结构上确定了 FSM 发挥作用的机制。通过正向遗传筛选,我们在两种人畜共患病葡萄球菌(施氏葡萄球菌和中间葡萄球菌)中鉴定出了促进 FSM 摄取的甘油-3-磷酸转运蛋白 GlpT。我们合成了一系列与 FSM 结构相关的亲脂性酯前药(称为 MEPicides),数据表明,前药部分的存在不仅显著提高了抑制剂对葡萄球菌的活性,而且还绕过了 GlpT 介导的细胞转运的需要。总的来说,我们的数据表明,前药 MEPicides 选择性且有效地抑制了人畜共患病葡萄球菌中的 DXR,并且 DXR 代表了未来开发的有前途的可成药靶点。