Capitanchik Charlotte, Toolan-Kerr Patrick, Luscombe Nicholas M, Ule Jernej
The Francis Crick Institute, London, United Kingdom.
Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology, London, United Kingdom.
Front Genet. 2020 May 20;11:398. doi: 10.3389/fgene.2020.00398. eCollection 2020.
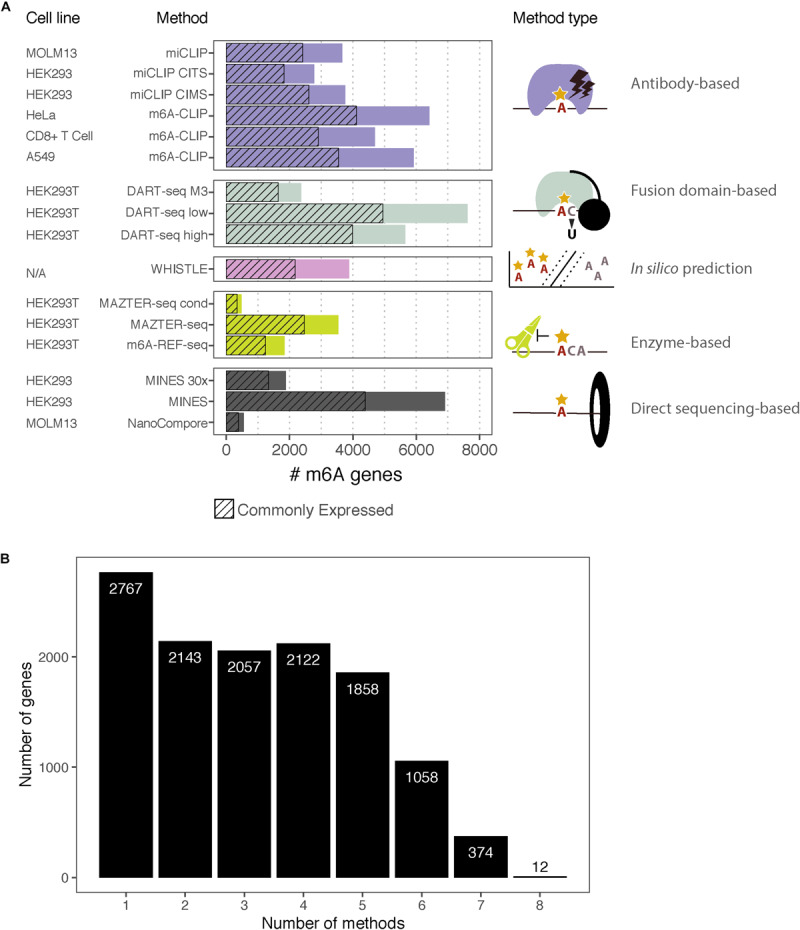
A flurry of methods has been developed in recent years to identify N6-methyladenosine (mA) sites across transcriptomes at high resolution. This raises the need to understand both the common features and those that are unique to each method. Here, we complement the analyses presented in the original papers by reviewing their various technical aspects and comparing the overlap between mA-methylated messenger RNAs (mRNAs) identified by each. Specifically, we examine eight different methods that identify mA sites in human cells with high resolution: two antibody-based crosslinking and immunoprecipitation (CLIP) approaches, two using endoribonuclease MazF, one based on deamination, two using Nanopore direct RNA sequencing, and finally, one based on computational predictions. We contrast the respective datasets and discuss the challenges in interpreting the overlap between them, including a prominent expression bias in detected genes. This overview will help guide researchers in making informed choices about using the available data and assist with the design of future experiments to expand our understanding of mA and its regulation.
近年来,已经开发出一系列方法,用于在全转录组范围内高分辨率地鉴定N6-甲基腺嘌呤(m⁶A)位点。这就需要我们了解这些方法的共同特征以及各自的独特之处。在这里,我们通过回顾各种技术方面并比较每种方法鉴定出的m⁶A甲基化信使核糖核酸(mRNA)之间的重叠,对原始论文中的分析进行补充。具体而言,我们研究了八种在人类细胞中高分辨率鉴定m⁶A位点的不同方法:两种基于抗体的交联免疫沉淀(CLIP)方法、两种使用核糖核酸内切酶MazF的方法、一种基于脱氨基作用的方法、两种使用纳米孔直接RNA测序的方法,以及最后一种基于计算预测的方法。我们对比了各自的数据集,并讨论了解释它们之间重叠所面临的挑战,包括检测到的基因中存在显著的表达偏差。本综述将有助于指导研究人员在使用现有数据时做出明智的选择,并协助设计未来的实验,以扩展我们对m⁶A及其调控的理解。