Li Chang, Zou Han, Xiong Zujian, Xiong Yi, Miyagishima Danielle F, Wanggou Siyi, Li Xuejun
Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha, China.
Xiangya School of Medicine, Central South University, Changsha, China.
Front Genet. 2020 May 19;11:429. doi: 10.3389/fgene.2020.00429. eCollection 2020.
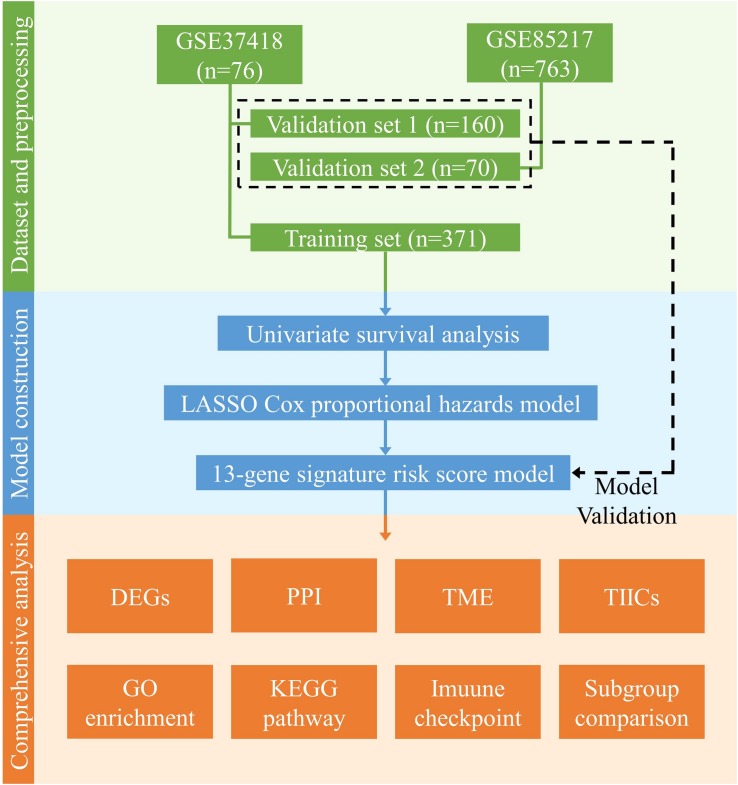
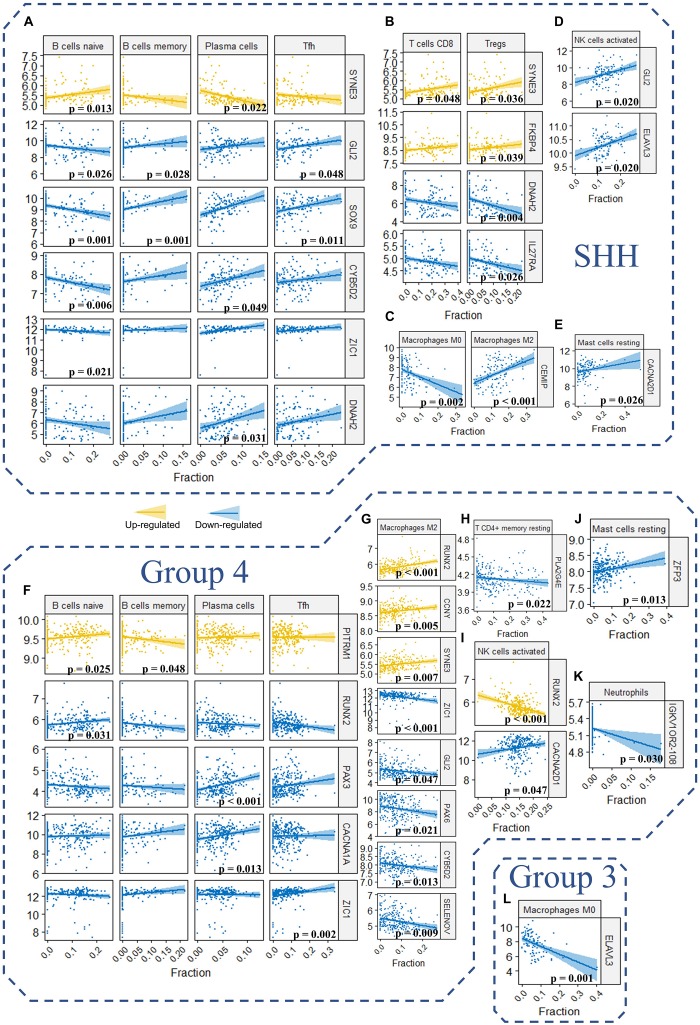
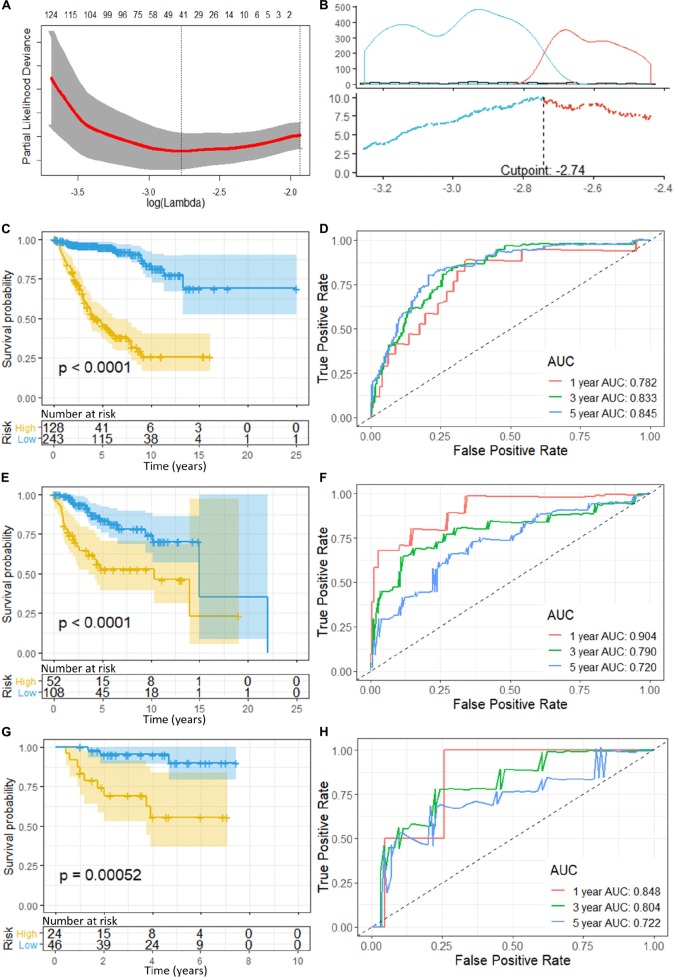
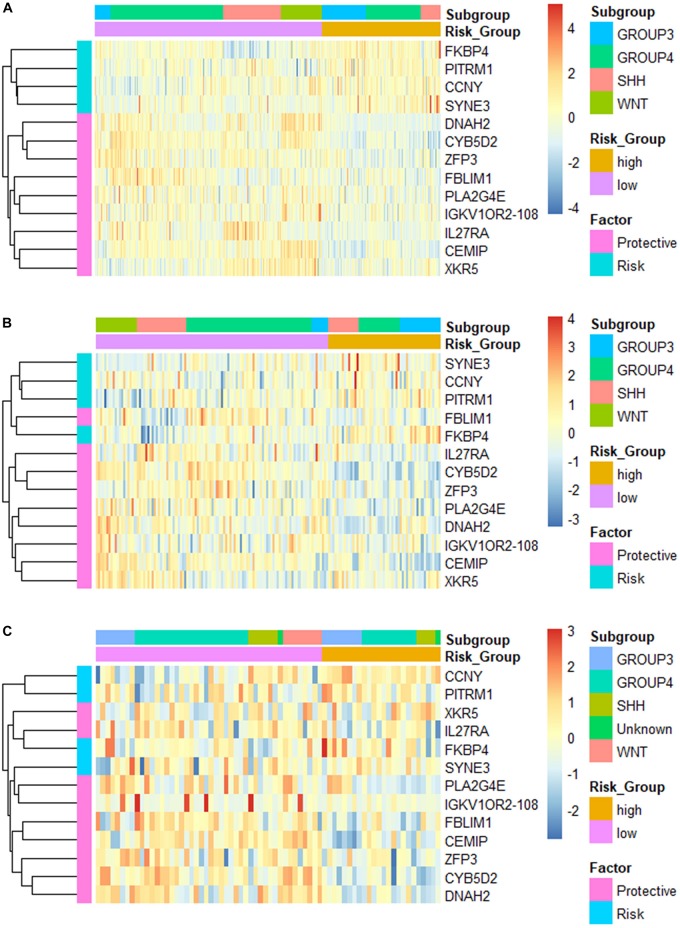
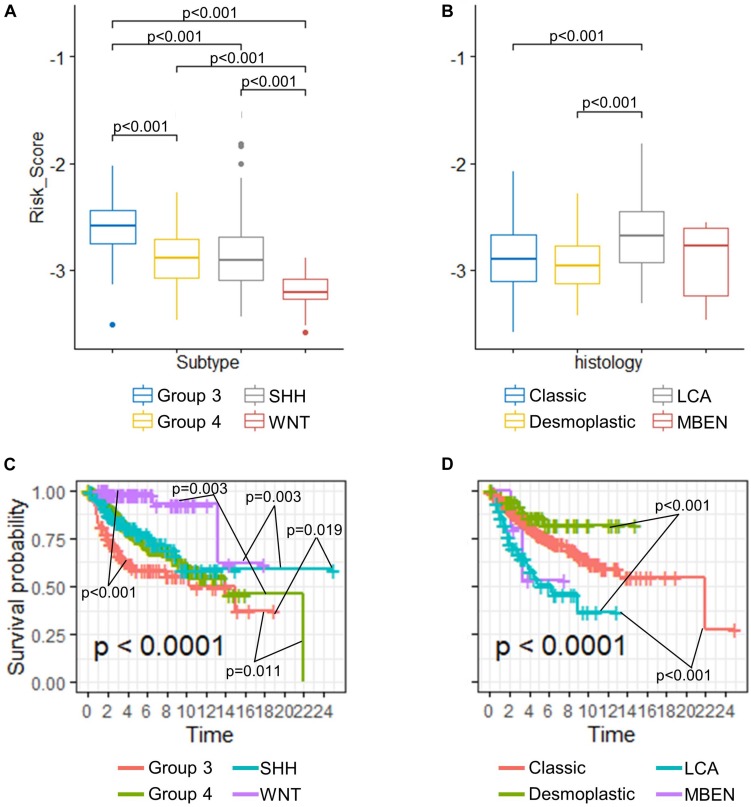
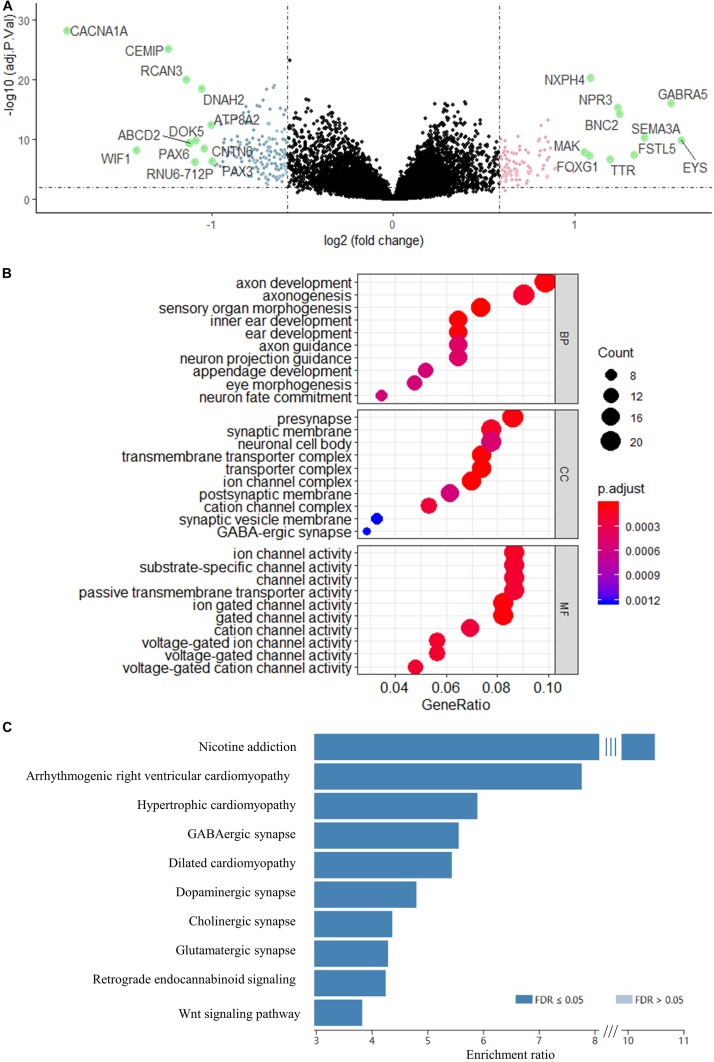
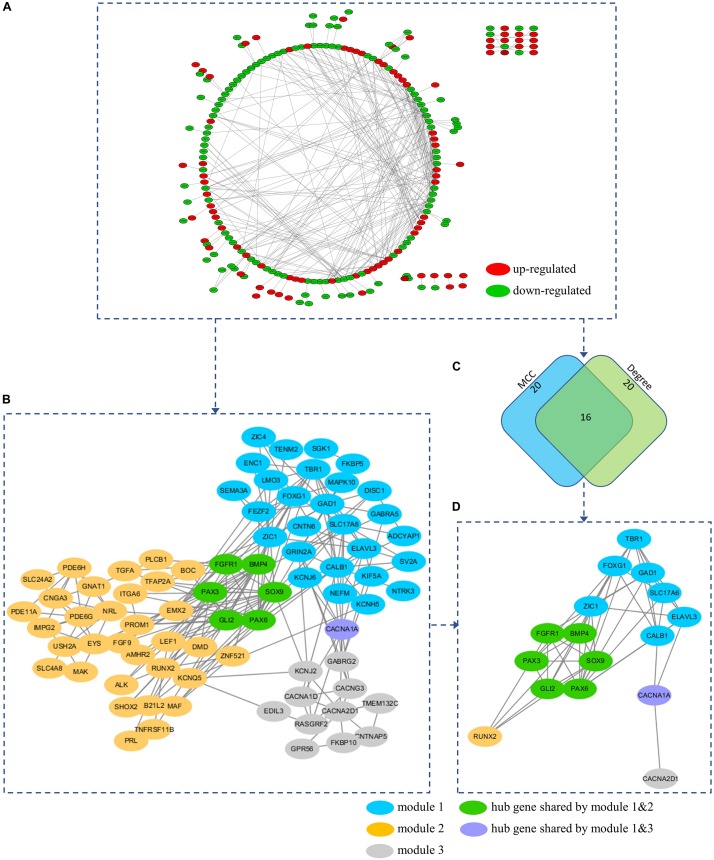
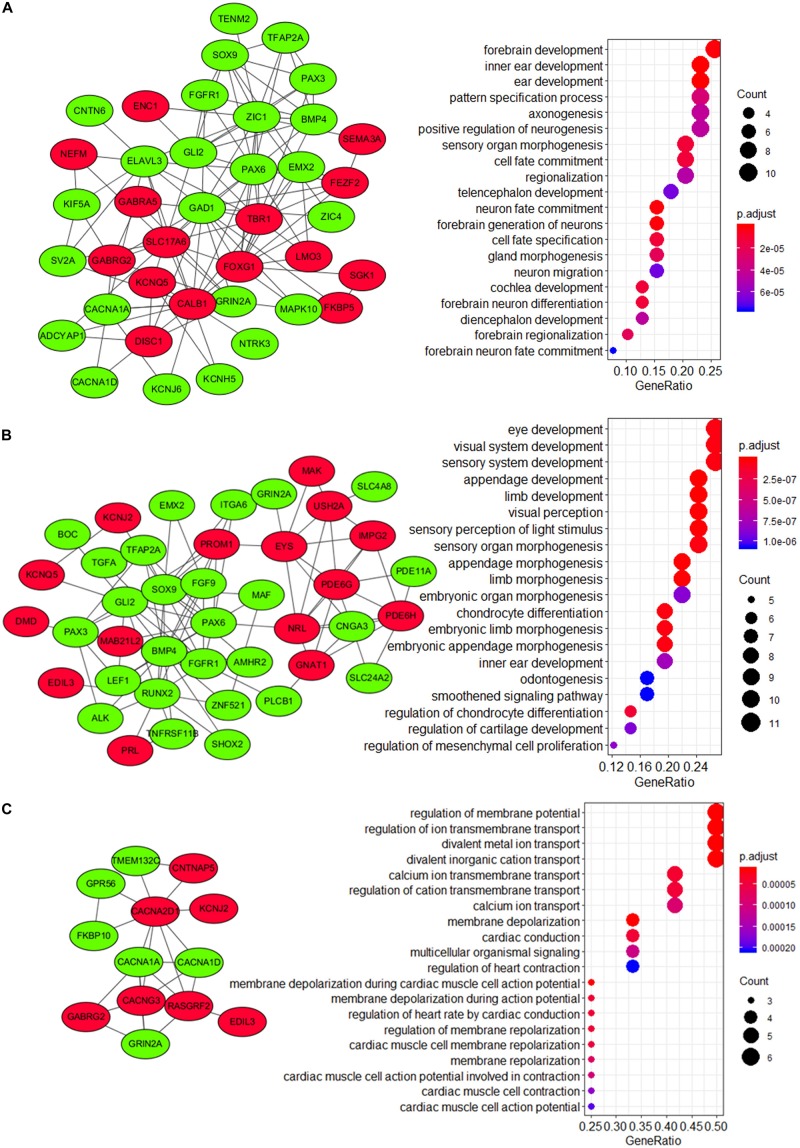
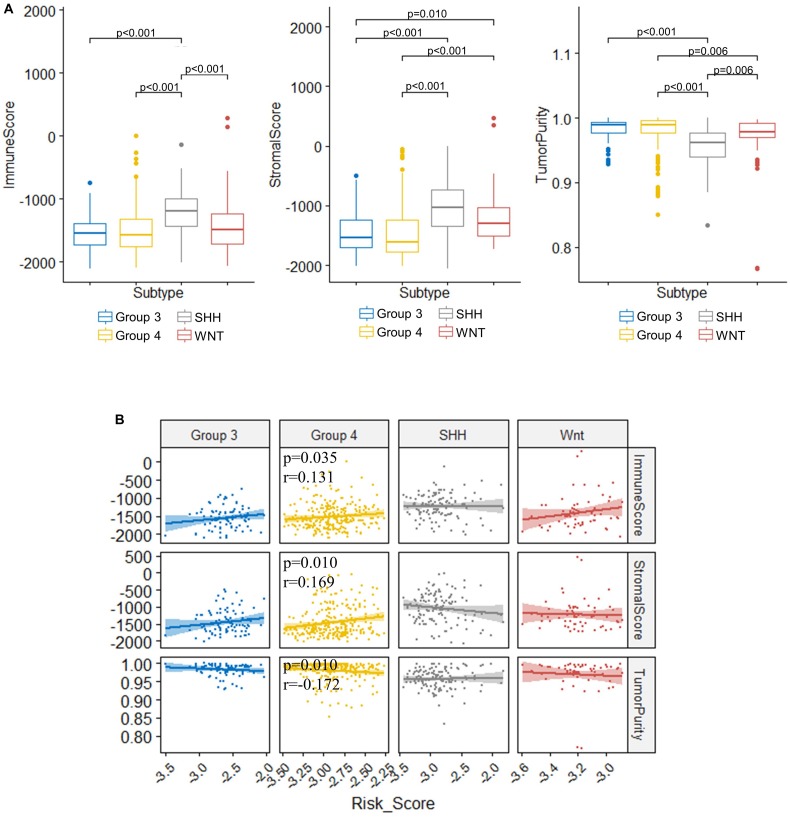
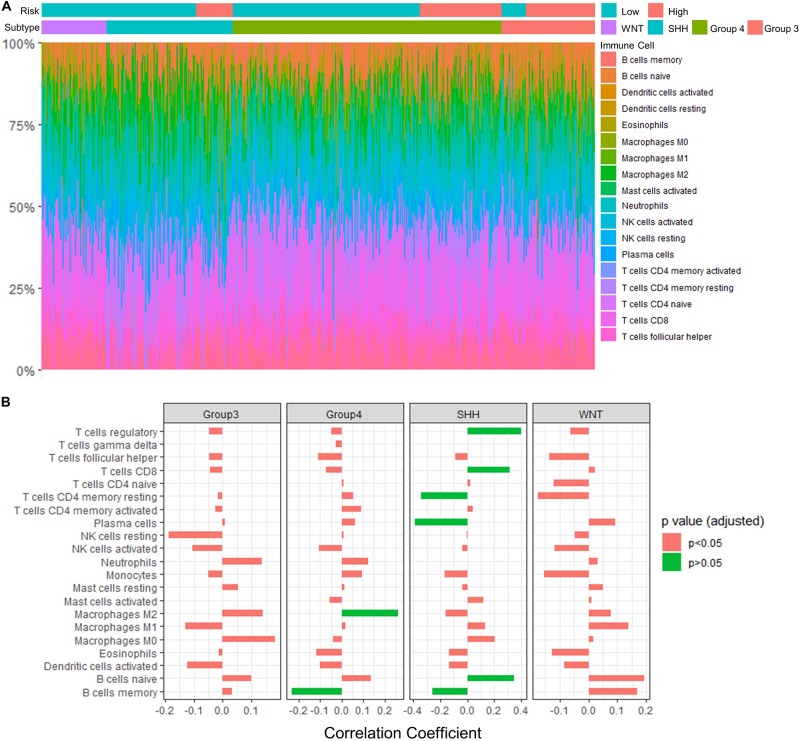
Recent studies have identified several molecular subgroups of medulloblastoma associated with distinct clinical outcomes; however, no robust gene signature has been established for prognosis prediction. Our objective was to construct a robust gene signature-based model to predict the prognosis of patients with medulloblastoma. Expression data of medulloblastomas were acquired from the Gene Expression Omnibus (GSE85217, = 763; GSE37418, = 76). To identify genes associated with overall survival (OS), we performed univariate survival analysis and least absolute shrinkage and selection operator (LASSO) Cox regression. A risk score model was constructed based on selected genes and was validated using multiple datasets. Differentially expressed genes (DEGs) between the risk groups were identified. Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), and protein-protein interaction (PPI) analyses were performed. Network modules and hub genes were identified using Cytoscape. Furthermore, tumor microenvironment (TME) was evaluated using ESTIMATE algorithm. Tumor-infiltrating immune cells (TIICs) were inferred using CIBERSORTx. A 13-gene model was constructed and validated. Patients classified as high-risk group had significantly worse OS than those as low-risk group (Training set: < 0.0001; Validation set 1: < 0.0001; Validation set 2: = 0.00052). The area under the curve (AUC) of the receiver operating characteristic (ROC) analysis indicated a good performance in predicting 1-, 3-, and 5-year OS in all datasets. Multivariate analysis integrating clinical factors demonstrated that the risk score was an independent predictor for the OS (validation set 1: = 0.001, validation set 2: = 0.004). We then identified 265 DEGs between risk groups and PPI analysis predicted modules that were highly related to central nervous system and embryonic development. The risk score was significantly correlated with programmed death-ligand 1 () expression ( < 0.001), as well as immune score ( = 0.035), stromal score ( = 0.010), and tumor purity ( = 0.010) in Group 4 medulloblastomas. Correlations between the 13-gene signature and the TIICs in Sonic hedgehog and Group 4 medulloblastomas were revealed. Our study constructed and validated a robust 13-gene signature model estimating the prognosis of medulloblastoma patients. We also revealed genes and pathways that may be related to the development and prognosis of medulloblastoma, which might provide candidate targets for future investigation.
近期研究已确定了髓母细胞瘤的几个分子亚组,这些亚组与不同的临床结局相关;然而,尚未建立用于预后预测的可靠基因特征。我们的目标是构建一个基于可靠基因特征的模型来预测髓母细胞瘤患者的预后。从基因表达综合数据库(GSE85217,n = 763;GSE37418,n = 76)获取髓母细胞瘤的表达数据。为了鉴定与总生存期(OS)相关的基因,我们进行了单变量生存分析和最小绝对收缩和选择算子(LASSO)Cox回归。基于选定基因构建了风险评分模型,并使用多个数据集进行了验证。确定了风险组之间的差异表达基因(DEG)。进行了京都基因与基因组百科全书(KEGG)、基因本体论(GO)和蛋白质 - 蛋白质相互作用(PPI)分析。使用Cytoscape鉴定了网络模块和枢纽基因。此外,使用ESTIMATE算法评估肿瘤微环境(TME)。使用CIBERSORTx推断肿瘤浸润免疫细胞(TIIC)。构建并验证了一个13基因模型。分类为高风险组的患者的OS明显比低风险组的患者差(训练集:P < 0.0001;验证集1:P < 0.0001;验证集2:P = 0.00052)。受试者操作特征(ROC)分析的曲线下面积(AUC)表明在所有数据集中预测1年、3年和5年OS方面表现良好。整合临床因素的多变量分析表明风险评分是OS的独立预测因子(验证集1:P = 0.001,验证集2:P = 0.004)。然后我们确定了风险组之间的265个DEG,PPI分析预测的模块与中枢神经系统和胚胎发育高度相关。在4组髓母细胞瘤中,风险评分与程序性死亡配体1(PD - L1)表达显著相关(P < 0.001),以及免疫评分(P = 0.035)、基质评分(P = 0.010)和肿瘤纯度(P = 0.010)。揭示了13基因特征与音猬因子型和4组髓母细胞瘤中的TIIC之间的相关性。我们的研究构建并验证了一个可靠的13基因特征模型,用于估计髓母细胞瘤患者的预后。我们还揭示了可能与髓母细胞瘤的发生和预后相关的基因和途径,这可能为未来的研究提供候选靶点。