Nasr Azadani Danial, Zhang Daiyuan, Hatherill J Robert, Silva David, Turner Jeffrey W
Life Sciences, Texas A&M University-Corpus Christi, Corpus Christi, TX, United States of America.
Natural Sciences, Del Mar College, Corpus Christi, TX, United States of America.
PeerJ. 2020 May 21;8:e9171. doi: 10.7717/peerj.9171. eCollection 2020.
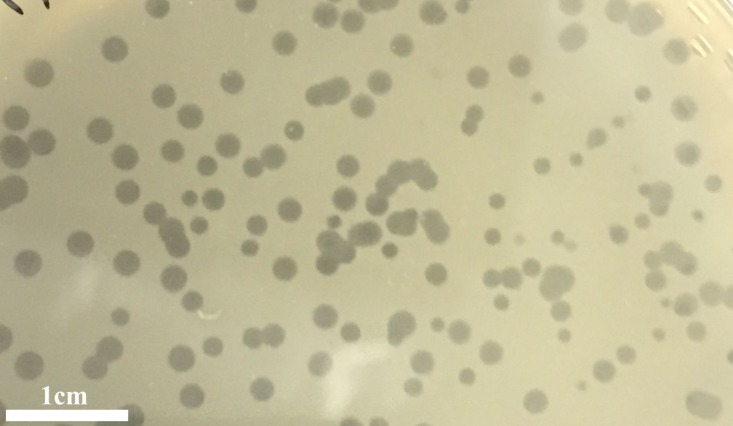
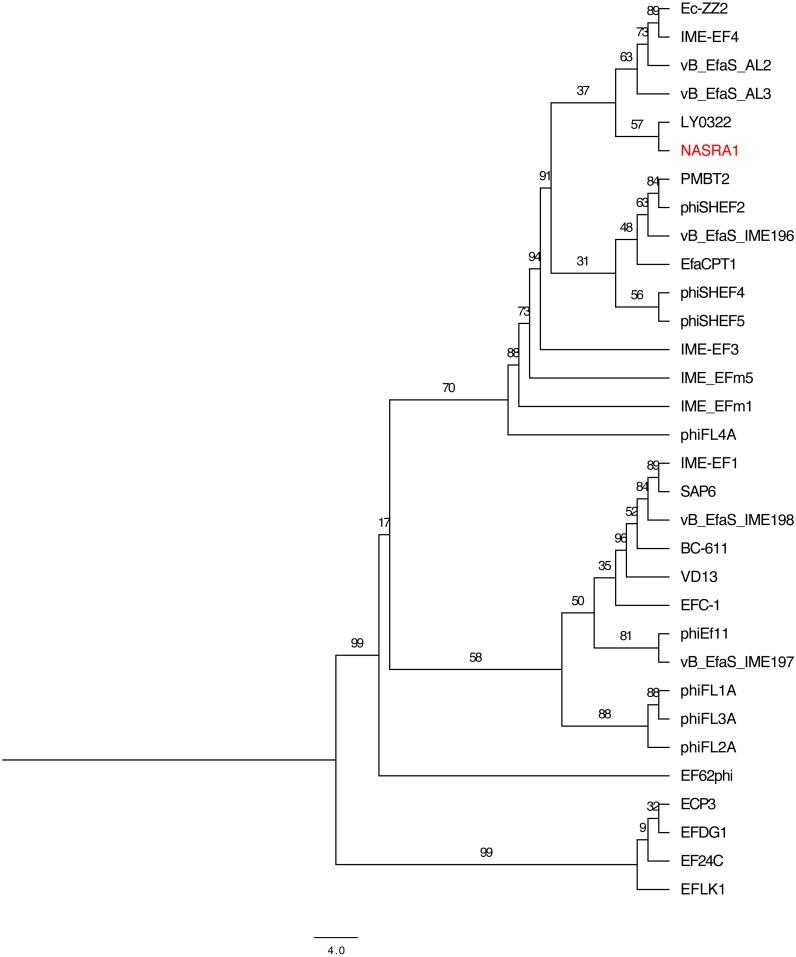
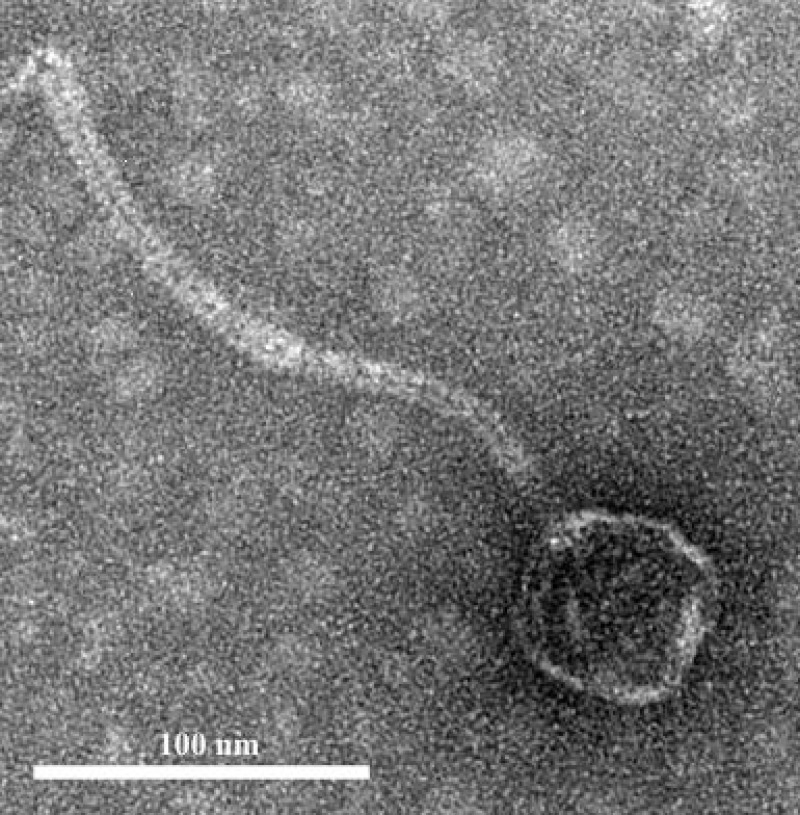
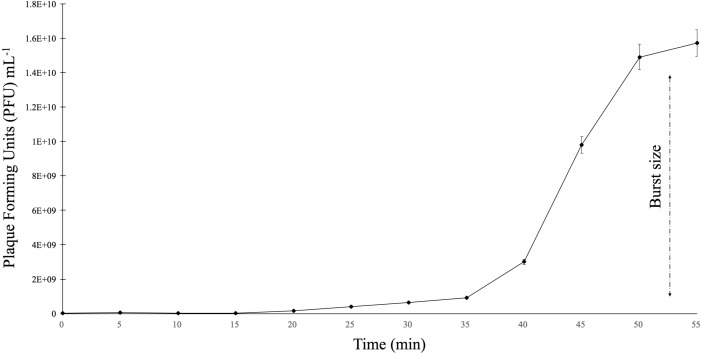
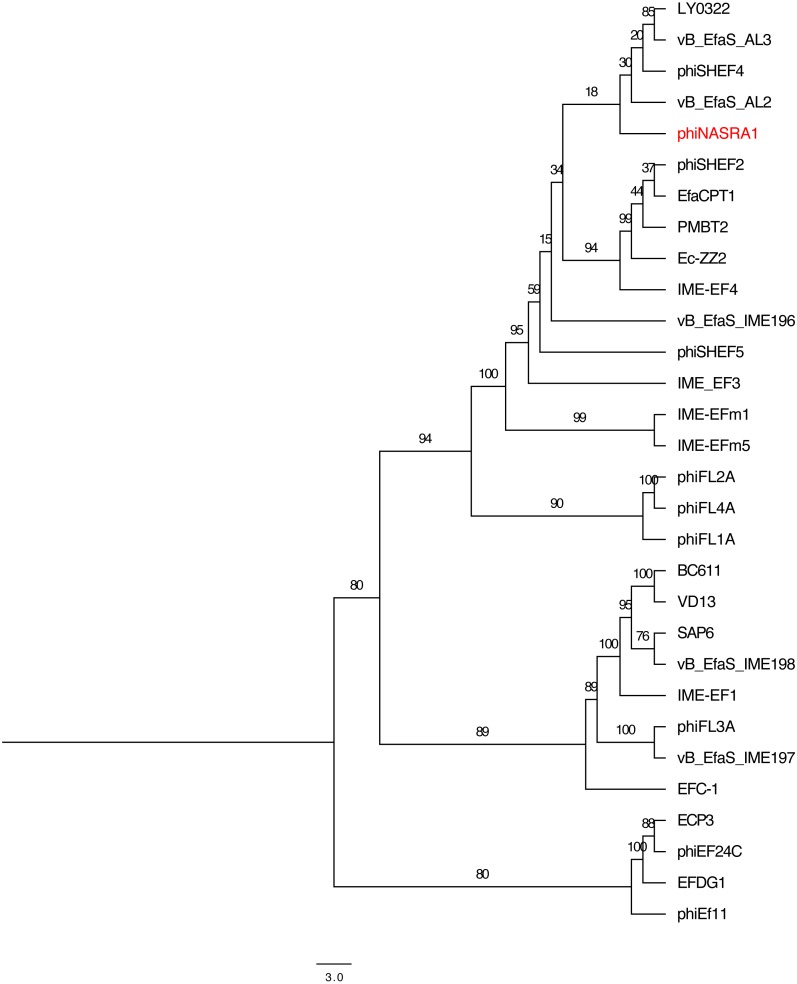
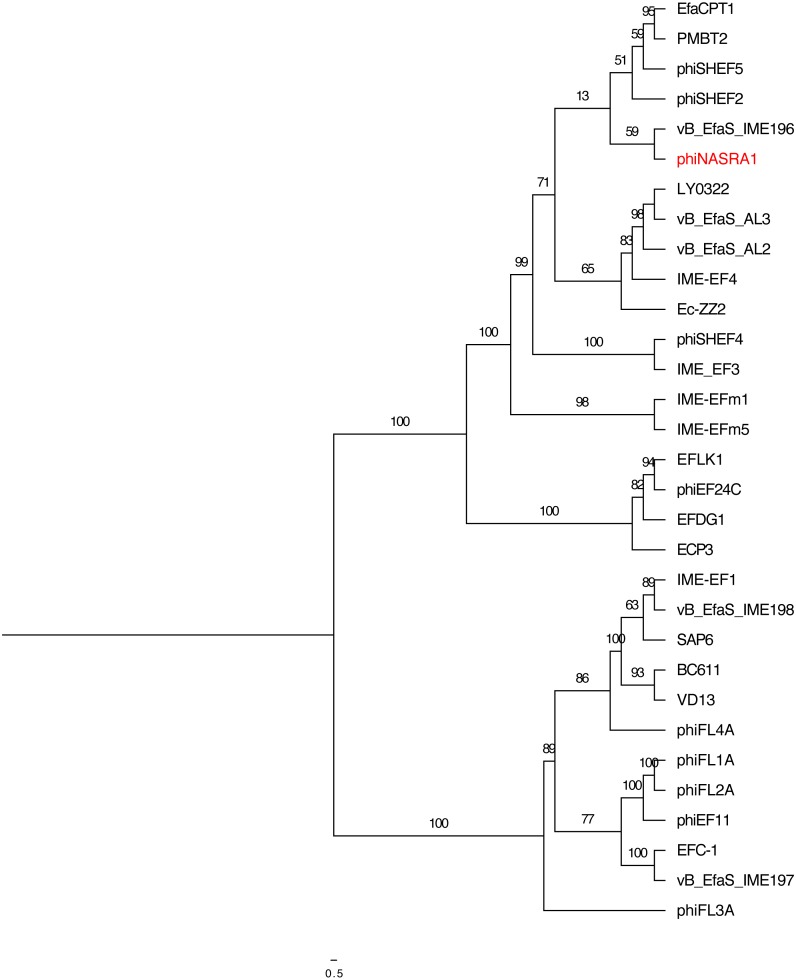
is a genus of Gram-positive bacteria that are commensal to the gastrointestinal tracts of humans but some species have been increasingly implicated as agents of nosocomial infections. The increase in infections and the spread of antibiotic-resistant strains have contributed to renewed interest in the discovery of phages. The aims of this study were (1) the isolation, characterization, and genome sequencing of a phage capable of infecting an antibiotic-resistant strain, and (2) the comparative genomic analysis of publicly-available phages. For this purpose, multiple phages were isolated from wastewater treatment plant (WWTP) influent using a high-level aminoglycoside-resistant (HLAR) strain as the host. One phage, phiNASRA1, demonstrated a high lytic efficiency (∼97.52%). Transmission electron microscopy (TEM) and whole-genome sequencing (WGS) showed that phiNASRA1 belongs to the Siphoviridae family of double-stranded DNA viruses. The phage was approximately 250 nm in length and its complete genome (40,139 bp, 34.7% GC) contained 62 open reading frames (ORFs). Phylogenetic comparisons of phiNASRA1 and 31 publicly-available phages, based on the large subunit terminase and portal proteins, grouped phage by provenance, size, and GC content. In particular, both phylogenies grouped phages larger than 100 kbp into distinct clades. A phylogeny based on a pangenome analysis of the same 32 phages also grouped phages by provenance, size, and GC content although agreement between the two single-locus phylogenies was higher. Per the pangenome phylogeny, phiNASRA1 was most closely related to phage LY0322 that was similar in size, GC content, and number of ORFs (40,139 and 40,934 bp, 34.77 and 34.80%, and 60 and 64 ORFs, respectively). The pangenome analysis did illustrate the high degree of sequence diversity and genome plasticity as no coding sequence was homologous across all 32 phages, and even 'conserved' structural proteins (e.g., the large subunit terminase and portal proteins) were homologous in no more than half of the 32 phage genomes. These findings contribute to a growing body of literature devoted to understanding phage biology and diversity. We propose that this high degree of diversity limited the value of the single-locus and pangenome phylogenies. By contrast, the high degree of homology between phages larger than 100 kbp suggests that pangenome analyses of more similar phages is a viable method for assessing subclade diversity. Future work is focused on validating phiNASRA1 as a potential therapeutic agent to eradicate antibiotic-resistant infections in an animal model.
是革兰氏阳性菌的一个属,它们是人类胃肠道的共生菌,但一些物种越来越多地被认为是医院感染的病原体。感染的增加和抗生素耐药菌株的传播促使人们对噬菌体的发现重新产生兴趣。本研究的目的是:(1)分离、表征和对能够感染抗生素耐药菌株的噬菌体进行基因组测序;(2)对公开可用的噬菌体进行比较基因组分析。为此,使用高水平氨基糖苷类耐药(HLAR)菌株作为宿主,从污水处理厂(WWTP)进水口分离出多种噬菌体。一种噬菌体phiNASRA1表现出高裂解效率(约97.52%)。透射电子显微镜(TEM)和全基因组测序(WGS)表明,phiNASRA1属于双链DNA病毒的长尾噬菌体科。该噬菌体长度约为250 nm,其完整基因组(40,139 bp,GC含量34.7%)包含62个开放阅读框(ORF)。基于大亚基末端酶和门户蛋白,对phiNASRA1和31个公开可用的噬菌体进行系统发育比较,按来源、大小和GC含量对噬菌体进行分组。特别是,两种系统发育分析都将大于100 kbp的噬菌体归为不同的进化枝。基于对相同32个噬菌体的泛基因组分析的系统发育分析也按来源、大小和GC含量对噬菌体进行分组,尽管两个单基因座系统发育分析之间的一致性更高。根据泛基因组系统发育分析,phiNASRA1与噬菌体LY0322关系最为密切,后者在大小、GC含量和ORF数量方面相似(分别为40,139和40,934 bp,34.77%和34.80%,以及60和64个ORF)。泛基因组分析确实说明了序列多样性和基因组可塑性的高度,因为在所有32个噬菌体中没有编码序列是同源的,甚至“保守”的结构蛋白(如大亚基末端酶和门户蛋白)在32个噬菌体基因组中同源性不超过一半。这些发现有助于增加致力于理解噬菌体生物学和多样性的文献数量。我们认为这种高度的多样性限制了单基因座和泛基因组系统发育分析的价值。相比之下,大于100 kbp的噬菌体之间的高度同源性表明,对更相似噬菌体的泛基因组分析是评估亚进化枝多样性的可行方法。未来的工作重点是验证phiNASRA1作为一种潜在治疗剂在动物模型中根除抗生素耐药感染的有效性。