Quadram Institute Biosciences, Norwich Research Park, Norwich, United Kingdom.
Earlham Institute, Norwich Research Park, Norwich, United Kingdom.
PLoS Genet. 2020 Jun 8;16(6):e1008850. doi: 10.1371/journal.pgen.1008850. eCollection 2020 Jun.
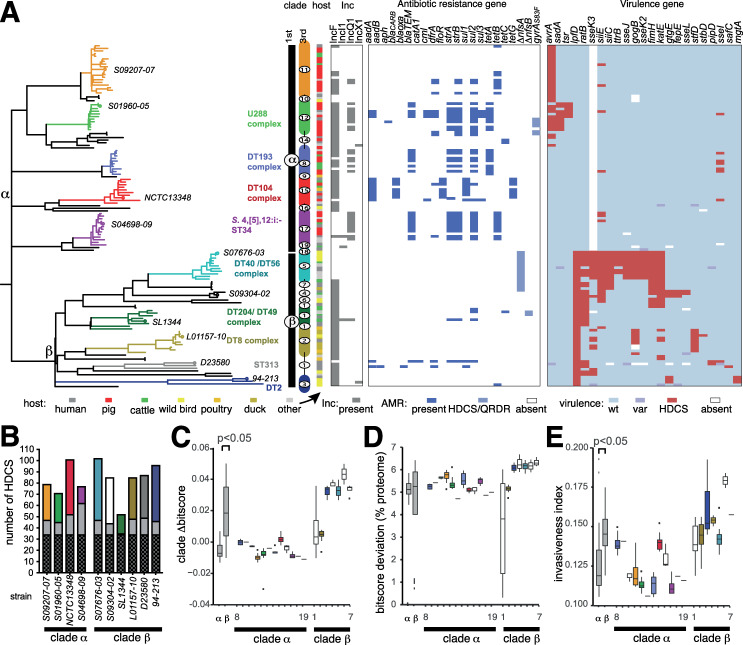
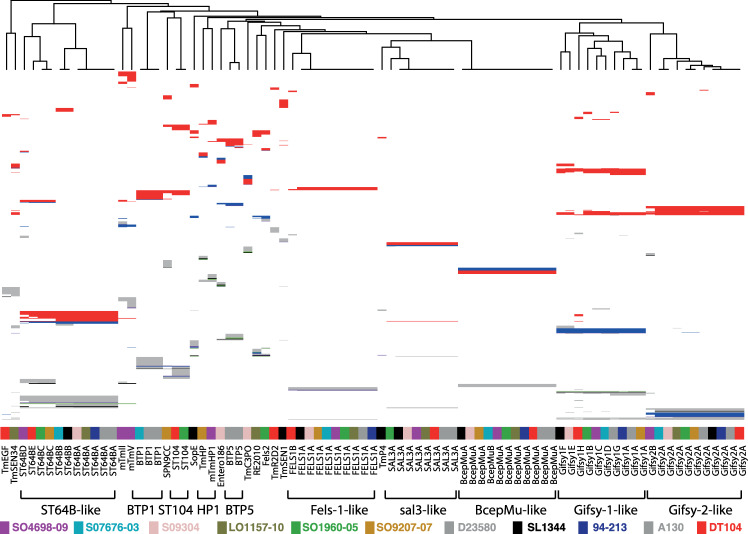
Salmonella enterica serotype Typhimurium (S. Typhimurium) is a leading cause of gastroenteritis and bacteraemia worldwide, and a model organism for the study of host-pathogen interactions. Two S. Typhimurium strains (SL1344 and ATCC14028) are widely used to study host-pathogen interactions, yet genotypic variation results in strains with diverse host range, pathogenicity and risk to food safety. The population structure of diverse strains of S. Typhimurium revealed a major phylogroup of predominantly sequence type 19 (ST19) and a minor phylogroup of ST36. The major phylogroup had a population structure with two high order clades (α and β) and multiple subclades on extended internal branches, that exhibited distinct signatures of host adaptation and anthropogenic selection. Clade α contained a number of subclades composed of strains from well characterized epidemics in domesticated animals, while clade β contained multiple subclades associated with wild avian species. The contrasting epidemiology of strains in clade α and β was reflected by the distinct distribution of antimicrobial resistance (AMR) genes, accumulation of hypothetically disrupted coding sequences (HDCS), and signatures of functional diversification. These observations were consistent with elevated anthropogenic selection of clade α lineages from adaptation to circulation in populations of domesticated livestock, and the predisposition of clade β lineages to undergo adaptation to an invasive lifestyle by a process of convergent evolution with of host adapted Salmonella serotypes. Gene flux was predominantly driven by acquisition and recombination of prophage and associated cargo genes, with only occasional loss of these elements. The acquisition of large chromosomally-encoded genetic islands was limited, but notably, a feature of two recent pandemic clones (DT104 and monophasic S. Typhimurium ST34) of clade α (SGI-1 and SGI-4).
鼠伤寒沙门氏菌(Salmonella enterica serotype Typhimurium,S. Typhimurium)是全球范围内导致胃肠炎和菌血症的主要原因,也是研究宿主-病原体相互作用的模式生物。两种鼠伤寒沙门氏菌菌株(SL1344 和 ATCC14028)被广泛用于研究宿主-病原体相互作用,但由于遗传变异,导致菌株具有不同的宿主范围、致病性和对食品安全的风险。不同鼠伤寒沙门氏菌菌株的种群结构揭示了一个主要的进化枝,主要由序列型 19(ST19)组成,还有一个较小的进化枝由 ST36 组成。主要进化枝具有两个高级阶元群(α 和 β)和多个扩展内部分支上的亚群,表现出明显的宿主适应性和人为选择的特征。α 进化枝包含多个由驯化动物中特征明确的流行菌株组成的亚群,而 β 进化枝包含与野生禽类物种相关的多个亚群。α 和 β 进化枝中菌株的对比流行病学反映在抗菌药物抗性(antimicrobial resistance,AMR)基因的不同分布、假设破坏的编码序列(hypothetically disrupted coding sequences,HDCS)的积累以及功能多样化的特征。这些观察结果与α 进化枝谱系在适应驯化动物种群循环中的人为选择增强以及β 进化枝谱系在通过与宿主适应的沙门氏菌血清型趋同进化的过程中发生适应性进化的倾向相一致。基因流动主要由噬菌体及其相关携带基因的获得和重组驱动,这些基因偶尔会丢失。染色体上大遗传岛的获得是有限的,但值得注意的是,α 进化枝的两个近期大流行克隆(DT104 和单相鼠伤寒沙门氏菌 ST34)具有 SGI-1 和 SGI-4。