Cai Jingwen, Drewry Michelle D, Perkumas Kristin, Dismuke W Michael, Hauser Michael A, Stamer W Daniel, Liu Yutao
Department of Cellular Biology and Anatomy, Medical College of Georgia, Augusta University, Augusta, GA.
Department of Ophthalmology, Duke University School of Medicine, Durham, NC.
Mol Vis. 2020 Jun 26;26:483-493. eCollection 2020.
Schlemm's canal (SC) endothelial cells derived from donors with or without glaucoma showed different mechanical properties and gene expression. As an important contributor to the regulation of intraocular pressure (IOP) and pathogenesis of primary open-angle glaucoma (POAG), the heritable key epigenetic changes, methylation may play an important role in the physiologic function of SC cells. This study aims to identify differentially methylated CpG sites (DMSs) in primary cultures of human SC cells with or without glaucoma.
We examined the methylation pattern of seven strains of primary human cells (two glaucoma and five normal SC cell samples), which were isolated and characterized using established protocols. DNA methylation was profiled using Illumina Human Methylation 450 BeadChip. Raw data were extracted and exported using Illumina GenomeStudio software. After quantile normalization, DNA methylation data were analyzed using R package RnBeads in Bioconductor. DMSs were filtered with p ≤ 1E-5, methylation change ≥ 0.1, and false discovery rate ≤ 0.05. The closest genes and the location of each CpG site were annotated using R package FDb.InfiniumMethylation.hg19. Gene Ontology and pathway analysis was performed using WebGestalt. Selected DMSs were validated using the Zymo qMethyl kit.
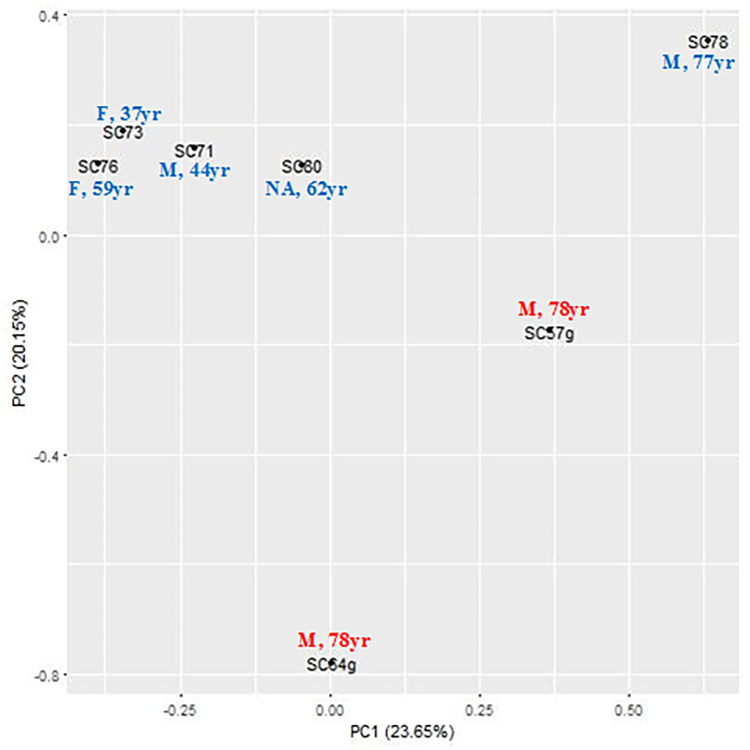
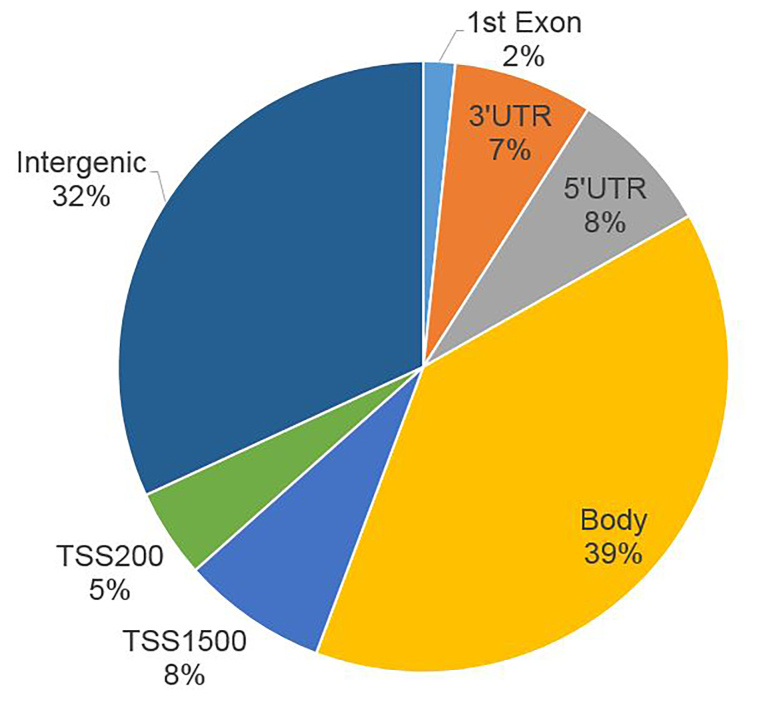
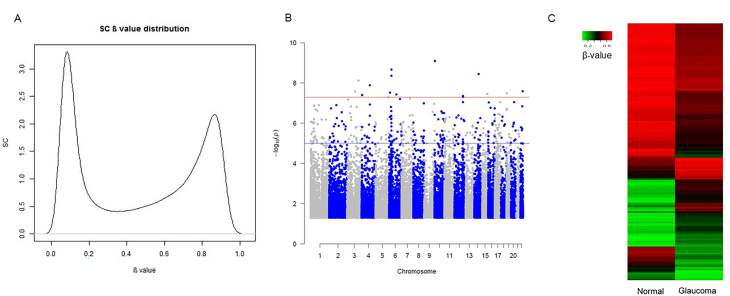
We used five non-glaucoma and two glaucomatous SC cell samples to profile genome-wide DNA methylation using Illumina Infinium Methylation BeadChips. Principle component analysis showed the separation between the glaucoma and control samples. After quality control and differential analysis, we identified 298 highly significant DMSs (p ≤ 1E-5). Among them, 221 DMSs were within 1 kb of a nearby gene. Gene Ontology analysis demonstrated significant enrichment in positive regulation of cell migration, negative regulation of endothelial cell proliferation, and stress fiber and actin filament bundles. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed enrichment in cell adhesion and gap junctions. Several glaucoma-related genes were identified, including , , , , , , , and . We also examined differentially methylated regions (DMRs) near these CpG sites and identified significant DMRs in , , and .
This study represents the first genome-wide DNA methylation profiling in cultured human primary SC cells. The DMSs were enriched in the pathways related to outflow resistance. Several DMRs were validated in glaucoma-associated genes, further suggesting the role of DNA methylation in glaucoma development. This study could provide comprehensive understanding of DNA methylation in glaucoma and its effect on aqueous humor outflow.
来自有或没有青光眼供体的施莱姆管(SC)内皮细胞表现出不同的机械性能和基因表达。作为眼内压(IOP)调节和原发性开角型青光眼(POAG)发病机制的重要贡献者,可遗传的关键表观遗传变化,即甲基化,可能在SC细胞的生理功能中起重要作用。本研究旨在鉴定有或没有青光眼的人SC细胞原代培养物中差异甲基化的CpG位点(DMS)。
我们检测了7株原代人细胞(2个青光眼和5个正常SC细胞样本)的甲基化模式,这些细胞使用既定方案分离和鉴定。使用Illumina Human Methylation 450 BeadChip对DNA甲基化进行分析。原始数据使用Illumina GenomeStudio软件提取和导出。经过分位数归一化后,使用Bioconductor中的R包RnBeads分析DNA甲基化数据。DMS以p≤1E-5、甲基化变化≥0.1和错误发现率≤0.05进行筛选。使用R包FDb.InfiniumMethylation.hg19注释每个CpG位点的最接近基因和位置。使用WebGestalt进行基因本体论和通路分析。使用Zymo qMethyl试剂盒验证选定的DMS。
我们使用5个非青光眼和2个青光眼SC细胞样本,使用Illumina Infinium Methylation BeadChips对全基因组DNA甲基化进行分析。主成分分析显示青光眼样本和对照样本之间的分离。经过质量控制和差异分析,我们鉴定出298个高度显著的DMS(p≤1E-5)。其中,221个DMS在附近基因的1 kb范围内。基因本体论分析表明在细胞迁移的正调控、内皮细胞增殖的负调控以及应力纤维和肌动蛋白丝束方面有显著富集。京都基因与基因组百科全书(KEGG)通路分析显示在细胞黏附和间隙连接方面有富集。鉴定出几个与青光眼相关的基因,包括 , , , , , , ,和 。我们还检查了这些CpG位点附近的差异甲基化区域(DMR),并在 , 和 中鉴定出显著的DMR。
本研究代表了首次在培养的人原代SC细胞中进行全基因组DNA甲基化分析。DMS在与房水流出阻力相关的通路中富集。在青光眼相关基因中验证了几个DMR,进一步表明DNA甲基化在青光眼发展中的作用。本研究可以提供对青光眼DNA甲基化及其对房水流出影响的全面理解。