Sayed Ahmed M, Alhadrami Hani A, El-Gendy Ahmed O, Shamikh Yara I, Belbahri Lassaad, Hassan Hossam M, Abdelmohsen Usama Ramadan, Rateb Mostafa E
Department of Pharmacognosy, Faculty of Pharmacy, Nahda University, 62513 Beni-Suef, Egypt.
Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, King Abdulaziz University, Jeddah 21589, Saudi Arabia.
Microorganisms. 2020 Jun 29;8(7):970. doi: 10.3390/microorganisms8070970.
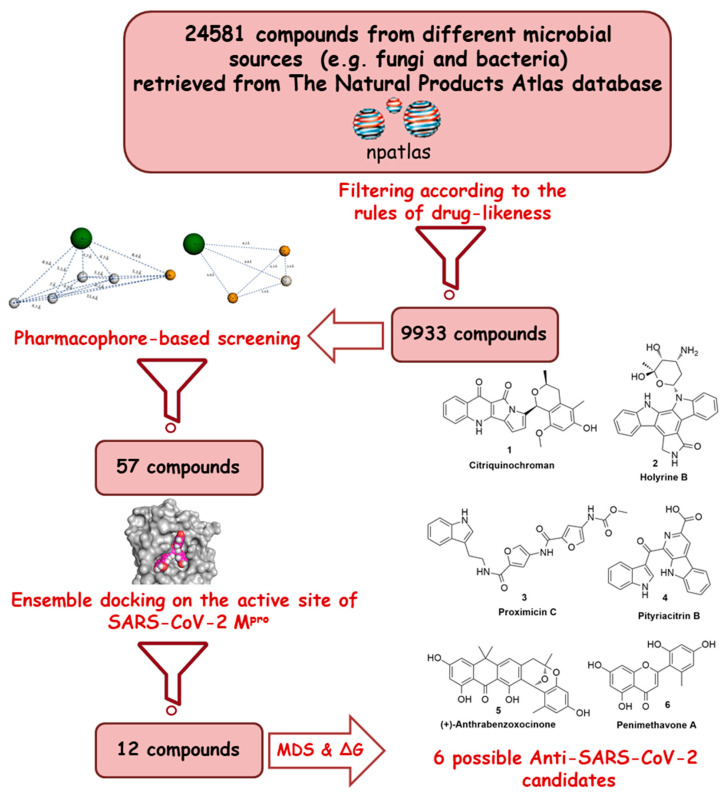
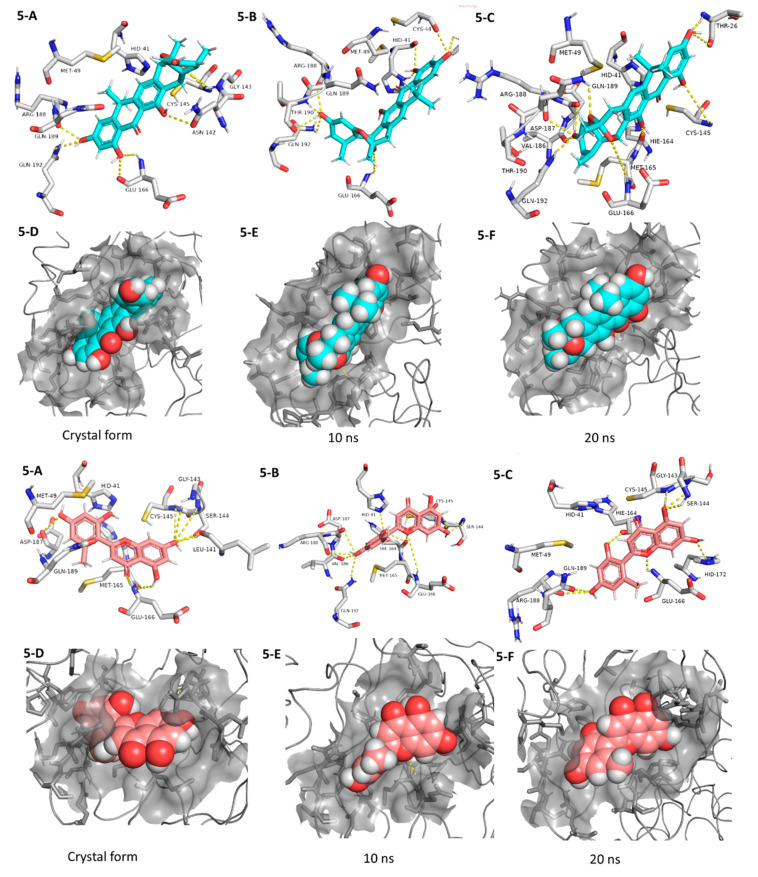
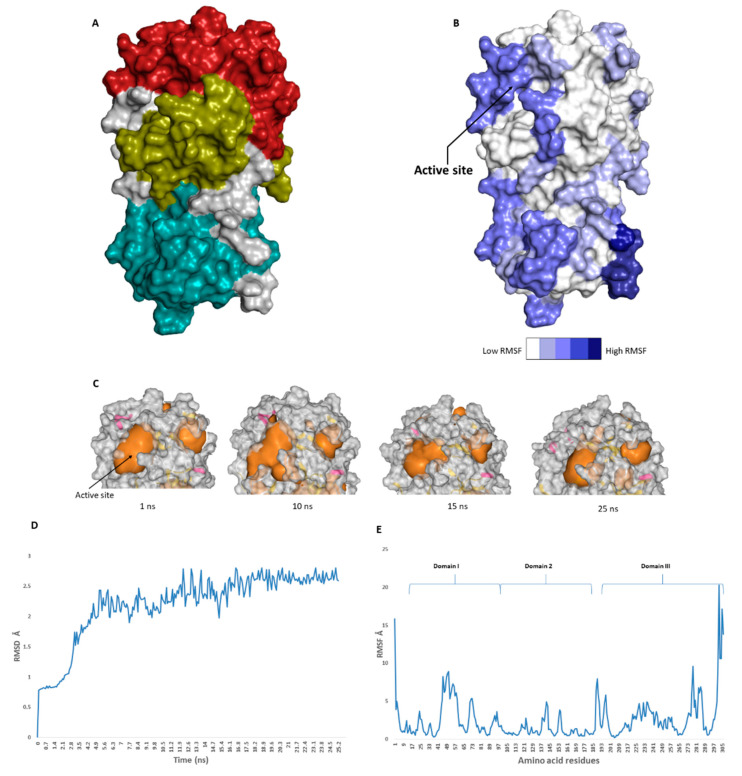
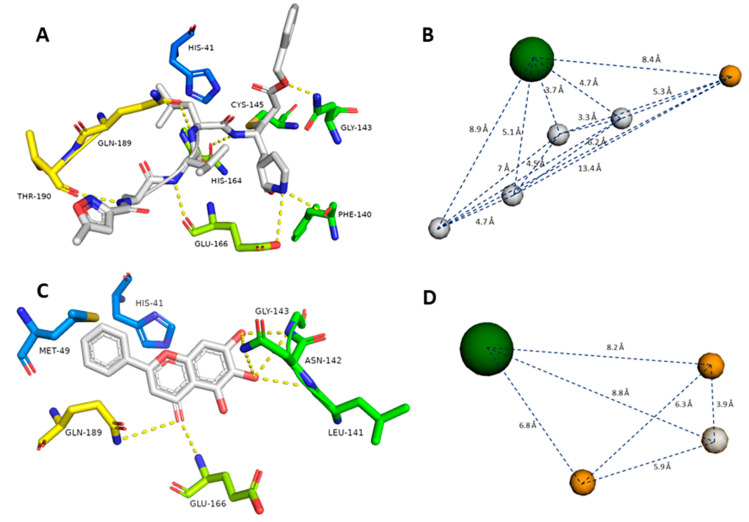
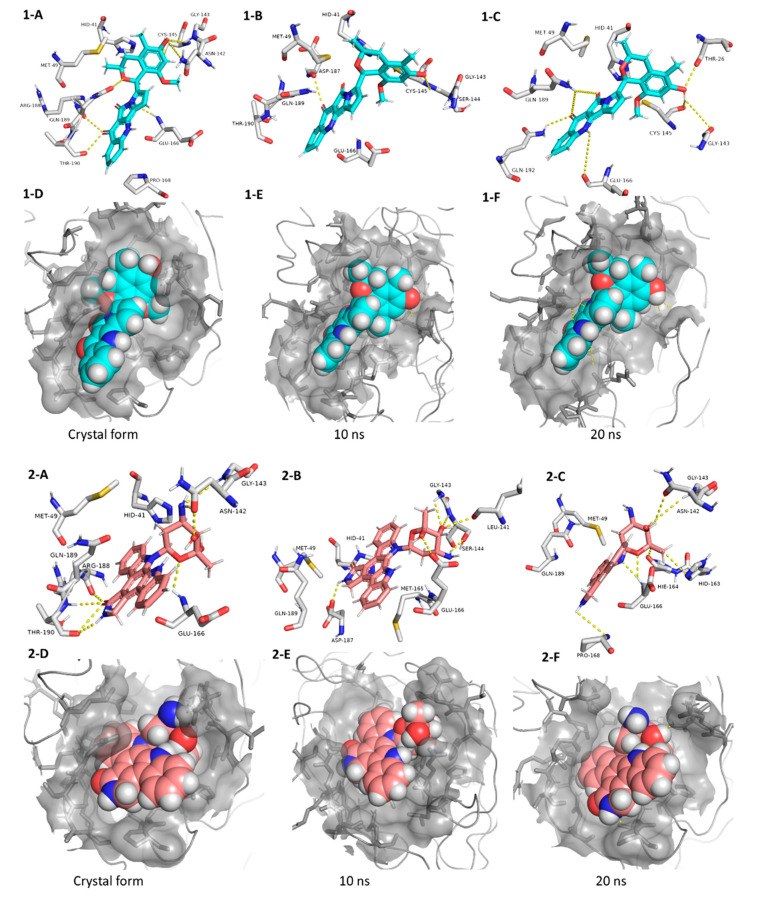
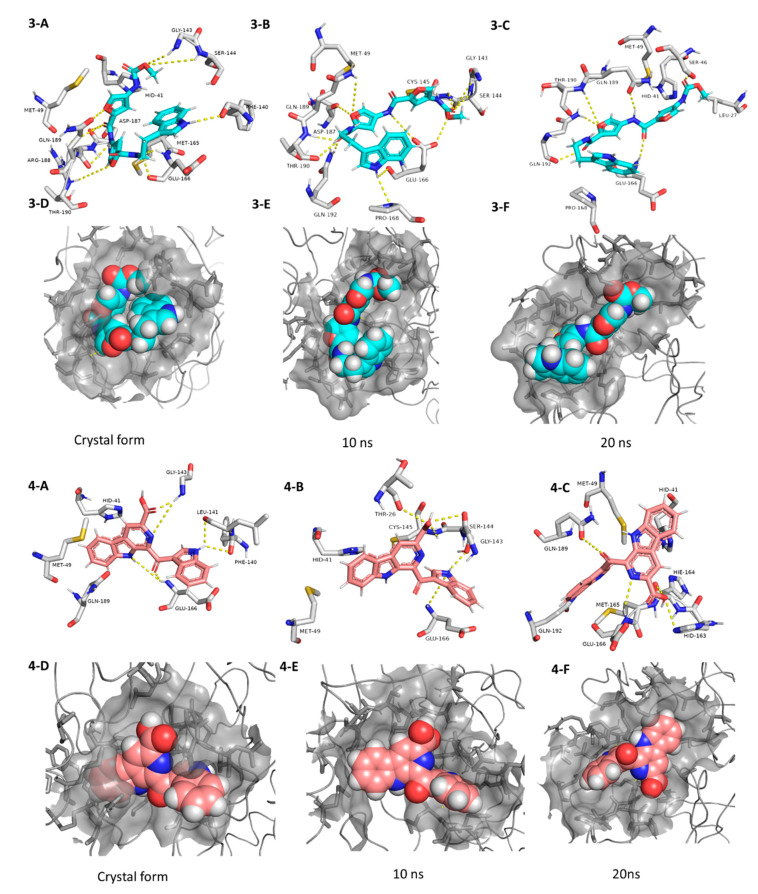
The main protease (M) of the newly emerged severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was subjected to hyphenated pharmacophoric-based and structural-based virtual screenings using a library of microbial natural products (>24,000 compounds). Subsequent filtering of the resulted hits according to the Lipinski's rules was applied to select only the drug-like molecules. Top-scoring hits were further filtered out depending on their ability to show constant good binding affinities towards the molecular dynamic simulation (MDS)-derived enzyme's conformers. Final MDS experiments were performed on the ligand-protein complexes (compounds Table S1) to verify their binding modes and calculate their binding free energy. Consequently, a final selection of six compounds () was proposed to possess high potential as anti-SARS-CoV-2 drug candidates. Our study provides insight into the role of the M structural flexibility during interactions with the possible inhibitors and sheds light on the structure-based design of anti-coronavirus disease 2019 (COVID-19) therapeutics targeting SARS-CoV-2.
利用一个微生物天然产物库(超过24000种化合物),对新出现的严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的主要蛋白酶(M)进行了基于药效团和基于结构的虚拟筛选联用。根据Lipinski规则对所得命中化合物进行后续筛选,以仅选择类药物分子。根据它们对分子动力学模拟(MDS)衍生的酶构象体显示持续良好结合亲和力的能力,进一步筛选得分最高的命中化合物。对配体-蛋白质复合物(化合物 表S1)进行最终的MDS实验,以验证它们的结合模式并计算它们的结合自由能。因此,最终选出六种化合物(),它们作为抗SARS-CoV-2药物候选物具有很高的潜力。我们的研究深入了解了M结构灵活性在与可能的抑制剂相互作用过程中的作用,并为针对SARS-CoV-2的2019冠状病毒病(COVID-19)治疗药物的基于结构的设计提供了线索。