California Institute for Quantitative Biosciences, University of California-Berkeley, Berkeley, California 94720, United States.
Department of Chemistry, Boston University, Boston, Massachusetts 02215, United States.
J Am Chem Soc. 2020 Jul 22;142(29):12620-12634. doi: 10.1021/jacs.0c02044. Epub 2020 Jul 9.
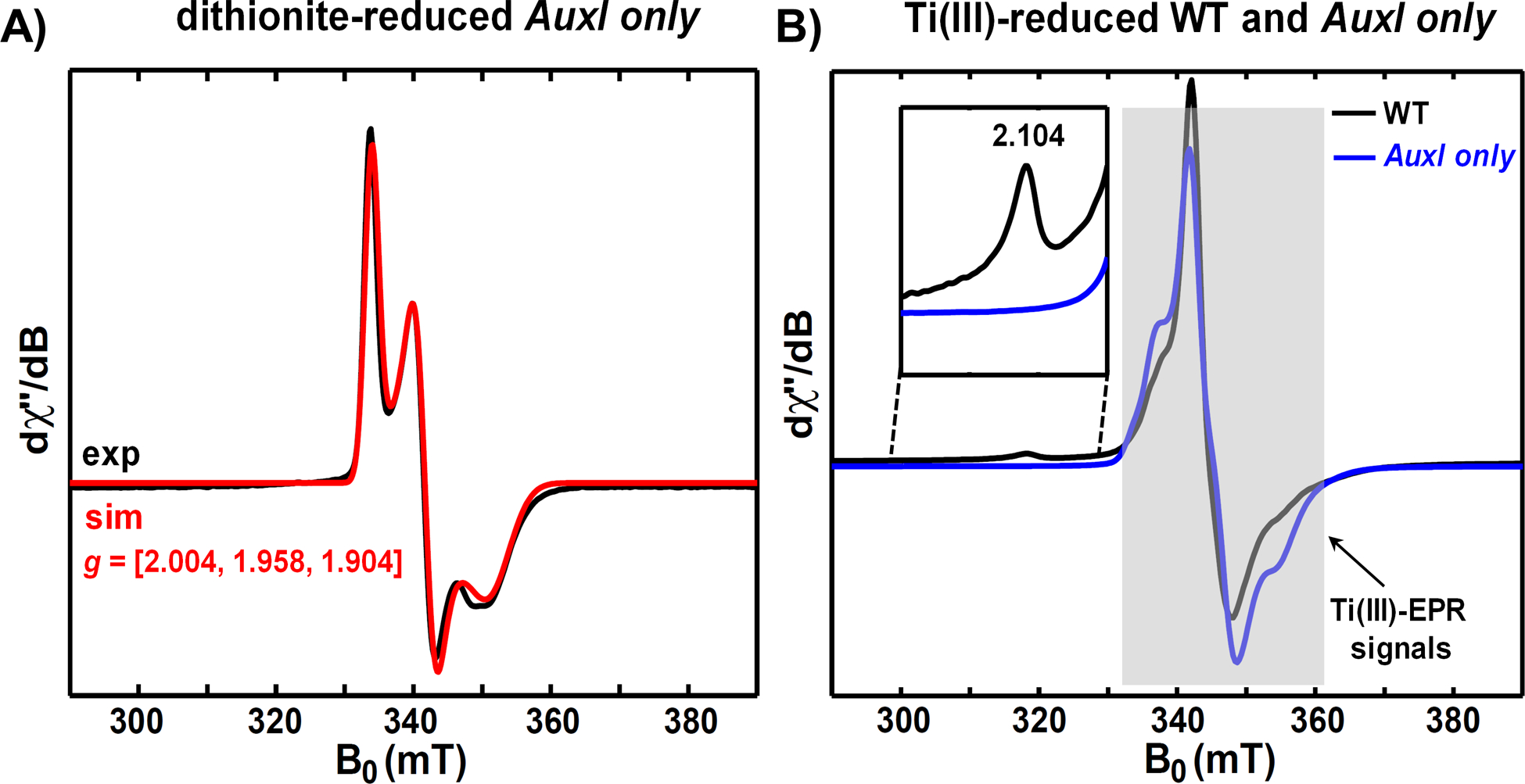
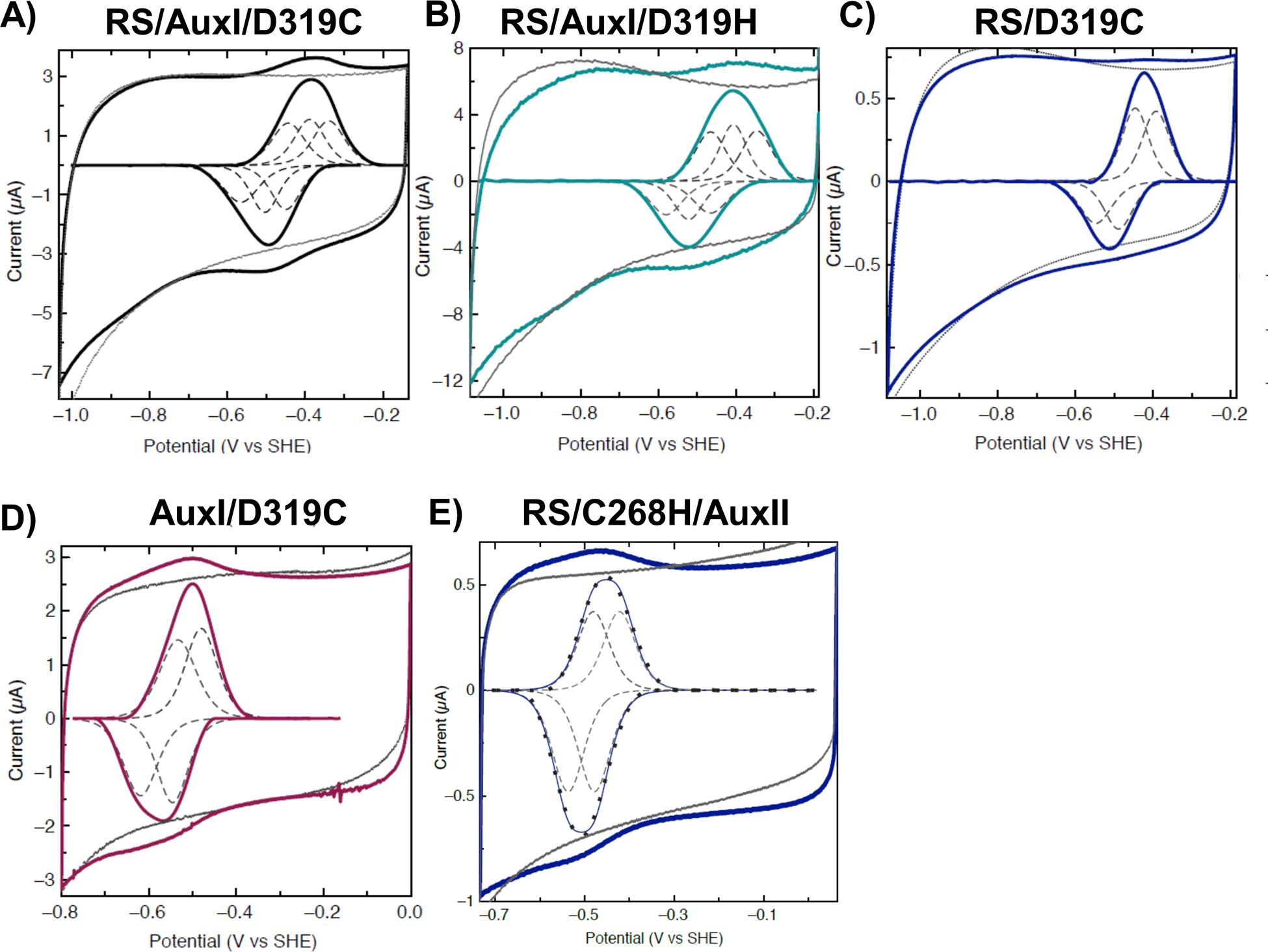
Understanding the relationship between the metallocofactor and its protein environment is the key to uncovering the mechanism of metalloenzymes. PqqE, a radical adenosylmethionine enzyme in pyrroloquinoline quinone (PQQ) biosynthesis, contains three iron-sulfur cluster binding sites. Two auxiliary iron-sulfur cluster binding sites, designated as AuxI and AuxII, use distinctive ligands compared to other proteins in the family while their functions remain unclear. Here, we investigate the electronic properties of these iron-sulfur clusters and compare the catalytic efficiency of wild-type (WT) AM1 PqqE to a range of mutated constructs. Using native mass spectrometry, protein film electrochemistry, and electron paramagnetic resonance spectroscopy, we confirm the previously proposed incorporation of a mixture of [2Fe-2S] and [4Fe-4S] clusters at the AuxI site and are able to assign redox potentials to each of the three iron-sulfur clusters. Significantly, a conservative mutation at AuxI, C268H, shown to selectively incorporate a [4Fe-4S] cluster, catalyzes an enhancement of uncoupled adenosylmethionine cleavage relative to WT, together with the elimination of detectable peptide cross-linked product. While a [4Fe-4S] cluster can be tolerated at the AuxI site, the aggregate findings suggest a functional [2Fe-2S] configuration within the AuxI site. PqqE variants with nondestructive ligand replacements at AuxII also show that the reduction potential at this site can be manipulated by changing the electronegativity of the unique aspartate ligand. A number of novel mechanistic features are proposed based on the kinetic and spectroscopic data. Additionally, bioinformatic analyses suggest that the unique ligand environment of PqqE may be relevant to its role in PQQ biosynthesis within an oxygen-dependent biosynthetic pathway.
理解金属辅因子与其蛋白质环境之间的关系是揭示金属酶机制的关键。PqqE 是吡咯喹啉醌 (PQQ) 生物合成中的一种自由基腺苷甲硫氨酸酶,它包含三个铁硫簇结合位点。两个辅助铁硫簇结合位点,分别称为 AuxI 和 AuxII,与家族中的其他蛋白质相比,使用独特的配体,但它们的功能仍不清楚。在这里,我们研究了这些铁硫簇的电子性质,并比较了野生型 (WT) AM1 PqqE 与一系列突变构建体的催化效率。使用天然质谱、蛋白质膜电化学和电子顺磁共振波谱,我们证实了先前提出的 AuxI 位点掺入[2Fe-2S]和[4Fe-4S]簇混合物的假设,并能够为三个铁硫簇中的每一个分配氧化还原电位。重要的是,AuxI 位点的保守突变 C268H 被证明选择性地掺入[4Fe-4S]簇,相对于 WT 催化未偶联的腺苷甲硫氨酸裂解增强,同时消除可检测的肽交联产物。虽然[4Fe-4S]簇可以在 AuxI 位点容忍,但综合研究结果表明 AuxI 位点存在功能性[2Fe-2S]构型。AuxII 位点具有非破坏性配体替换的 PqqE 变体也表明,通过改变独特天冬氨酸配体的电负性,可以操纵该位点的还原电位。根据动力学和光谱数据提出了许多新的机制特征。此外,生物信息学分析表明,PqqE 的独特配体环境可能与其在依赖氧的生物合成途径中参与 PQQ 生物合成的作用有关。