Sullivan Sandra S, Weinzierl Robert O J
Department of Life Sciences, Imperial College London, London SW7 2AZ, UK.
Life (Basel). 2020 Jul 10;10(7):109. doi: 10.3390/life10070109.
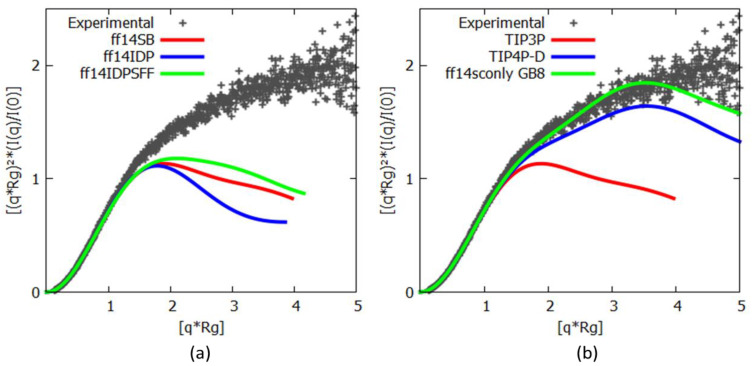
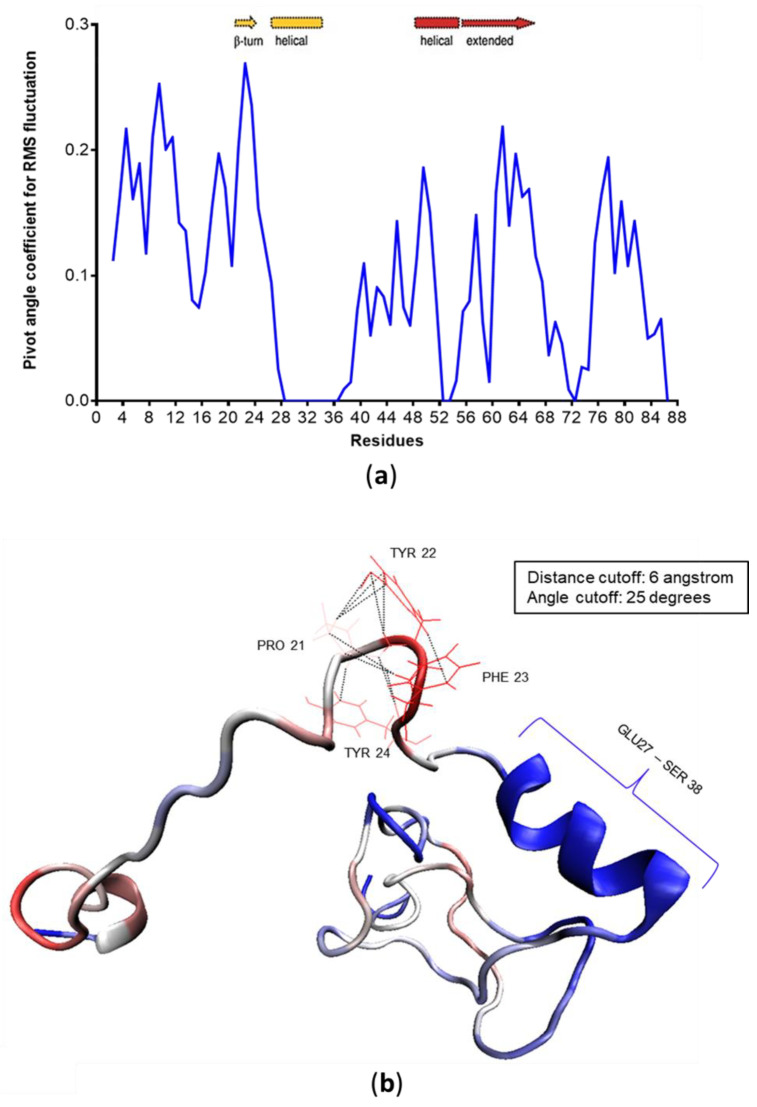
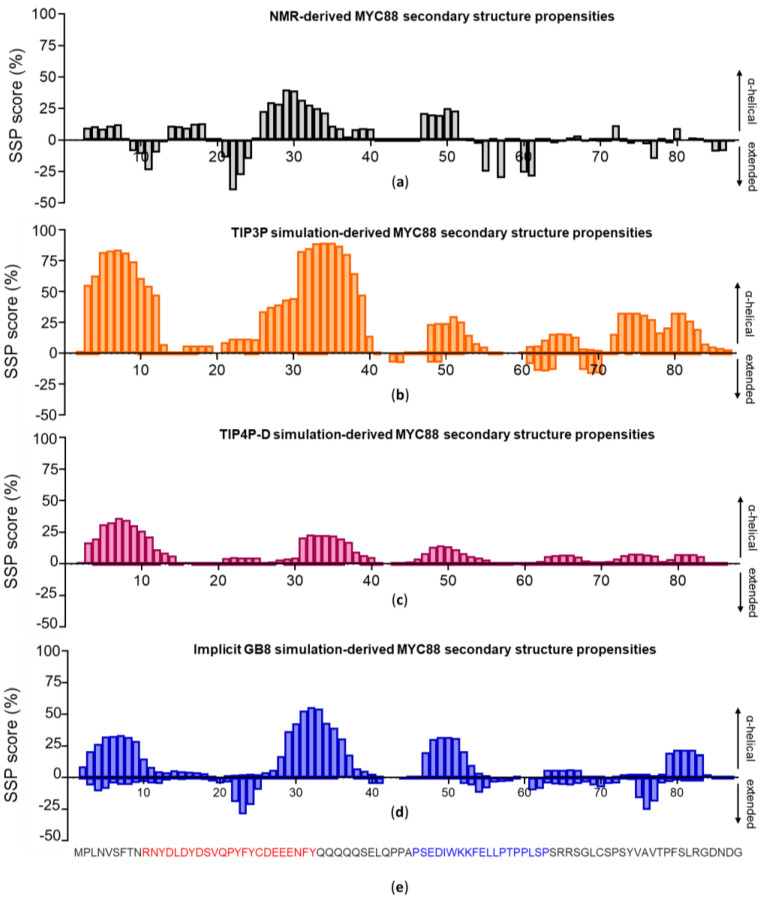
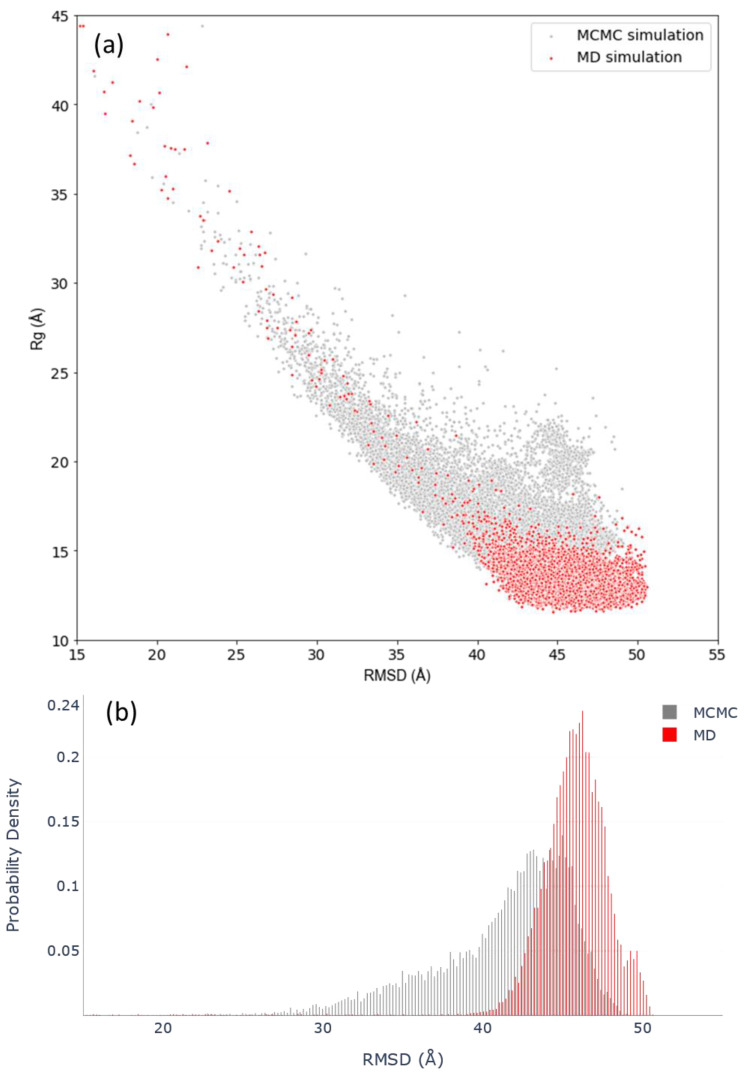
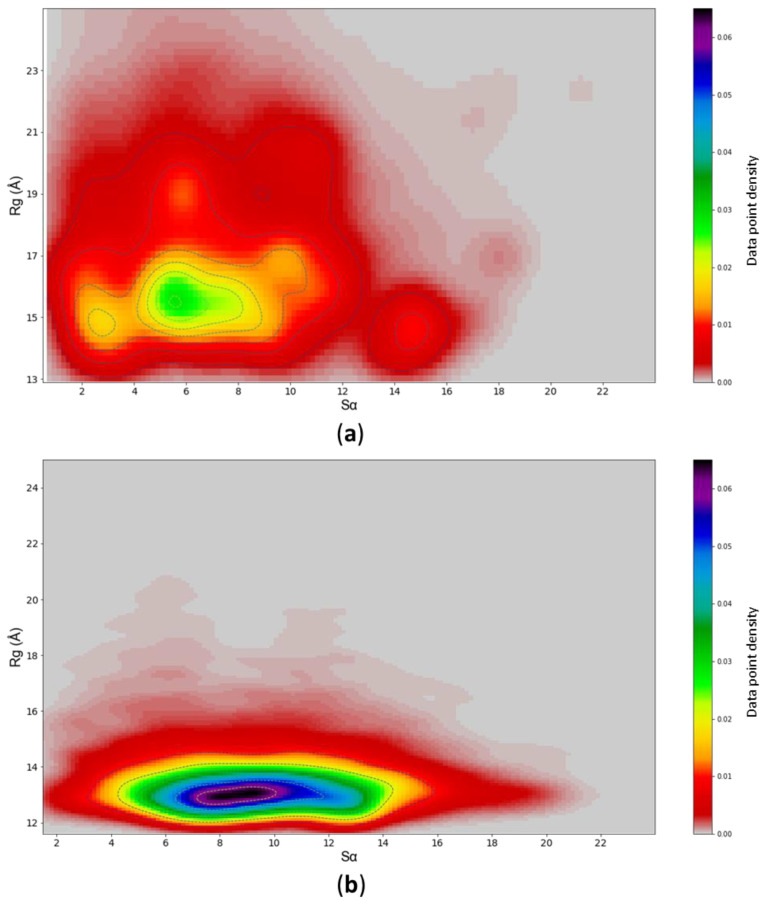
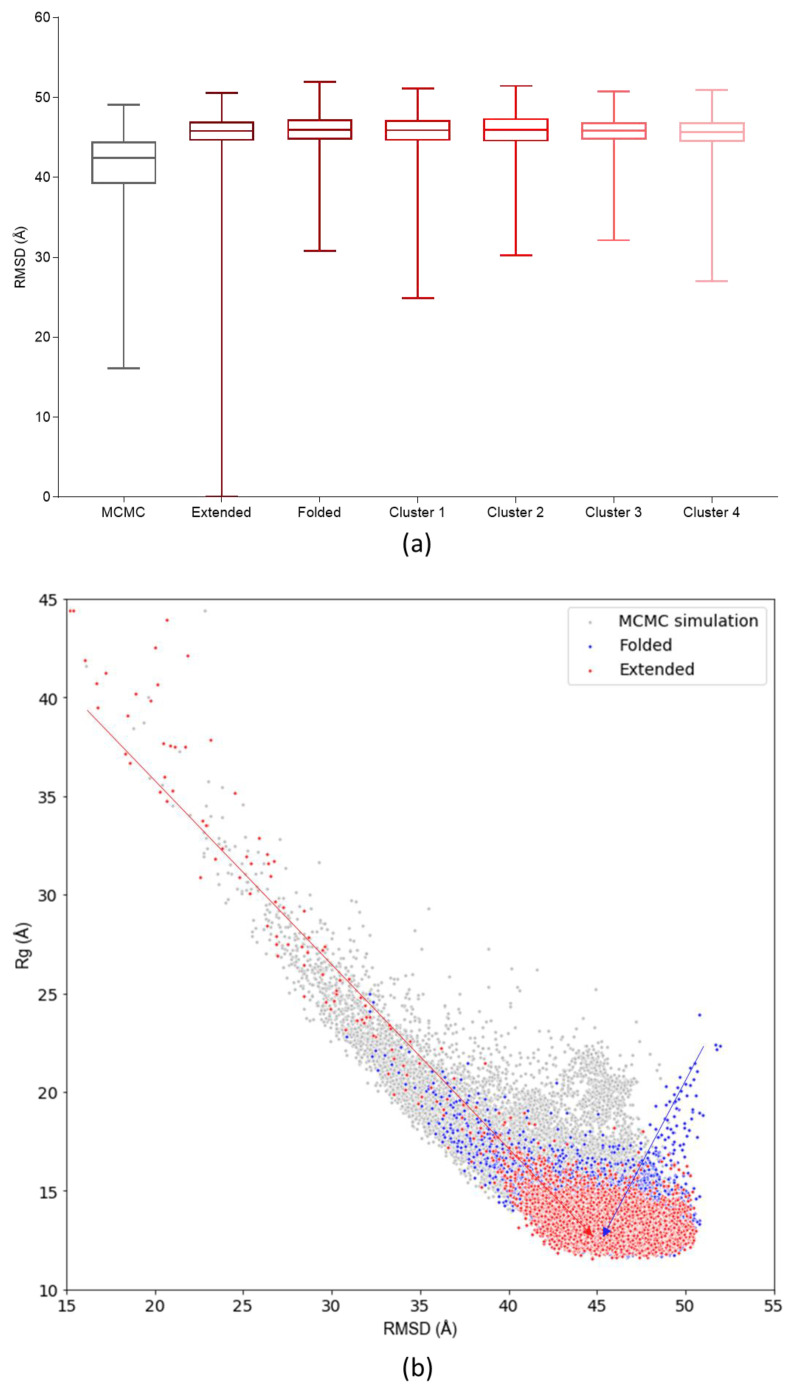
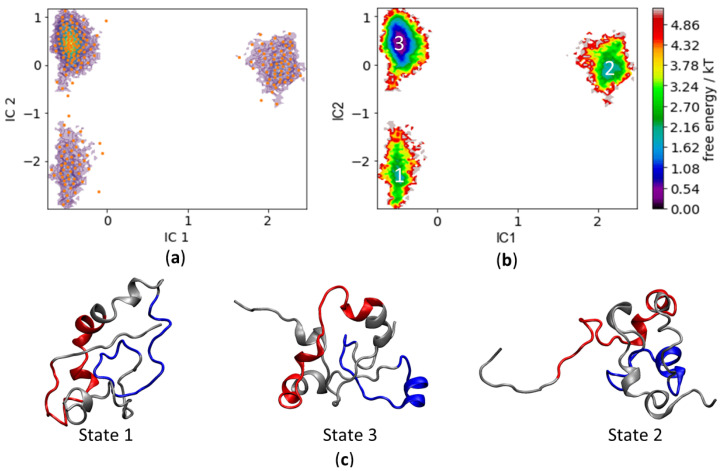
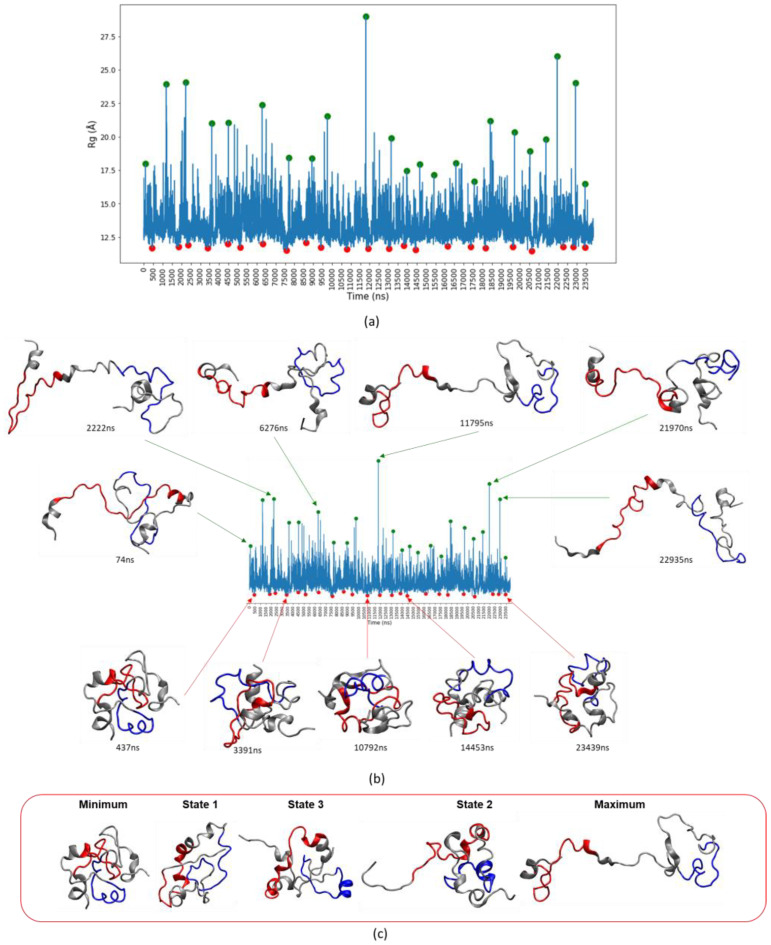
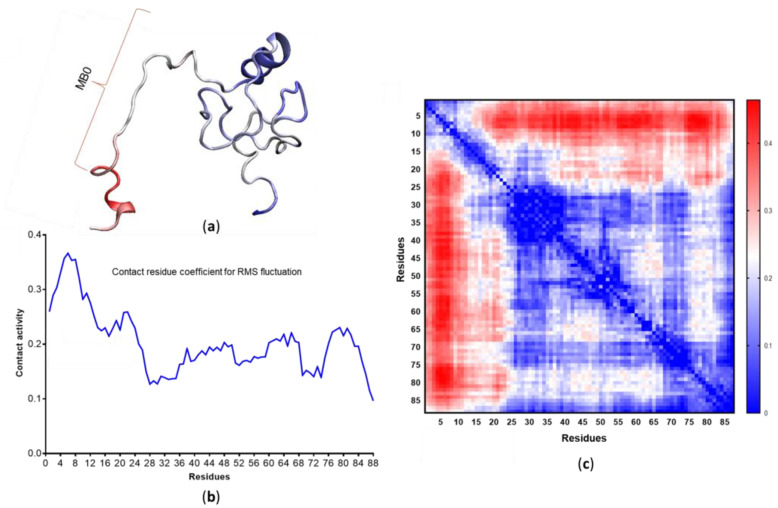
Many of the proteins involved in key cellular regulatory events contain extensive intrinsically disordered regions that are not readily amenable to conventional structure/function dissection. The oncoprotein c-MYC plays a key role in controlling cell proliferation and apoptosis and more than 70% of the primary sequence is disordered. Computational approaches that shed light on the range of secondary and tertiary structural conformations therefore provide the only realistic chance to study such proteins. Here, we describe the results of several tests of force fields and water models employed in molecular dynamics simulations for the N-terminal 88 amino acids of c-MYC. Comparisons of the simulation data with experimental secondary structure assignments obtained by NMR establish a particular implicit solvation approach as highly congruent. The results provide insights into the structural dynamics of c-MYC, which will be useful for guiding future experimental approaches. The protocols for trajectory analysis described here will be applicable for the analysis of a variety of computational simulations of intrinsically disordered proteins.
许多参与关键细胞调控事件的蛋白质都含有广泛的内在无序区域,这些区域不易用传统的结构/功能剖析方法进行研究。癌蛋白c-MYC在控制细胞增殖和凋亡中起关键作用,其一级序列的70%以上是无序的。因此,能够揭示二级和三级结构构象范围的计算方法为研究这类蛋白质提供了唯一现实的机会。在这里,我们描述了对c-MYC N端88个氨基酸进行分子动力学模拟时所采用的几种力场和水模型的测试结果。将模拟数据与通过核磁共振获得的实验二级结构归属进行比较,确定了一种高度一致的特定隐式溶剂化方法。这些结果为c-MYC的结构动力学提供了见解,这将有助于指导未来的实验方法。这里描述的轨迹分析协议将适用于分析各种内在无序蛋白质的计算模拟。