Institute of Ecology and Evolution, University of Bern, 3012 Bern, Switzerland.
Swiss Institute of Bioinformatics, Quartier Sorge, 1011 Lausanne, Switzerland.
Viruses. 2020 Jul 11;12(7):749. doi: 10.3390/v12070749.
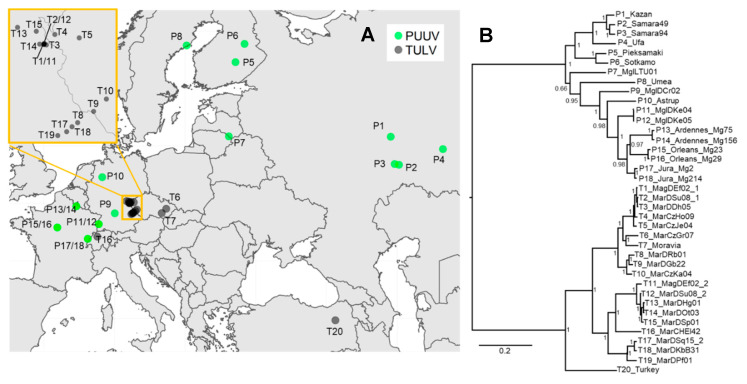
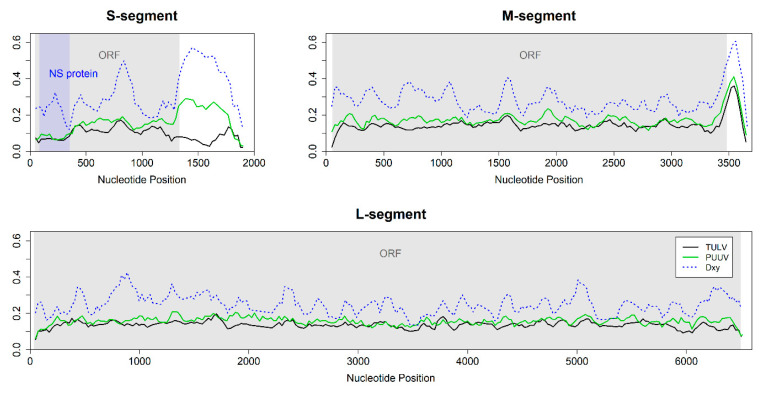
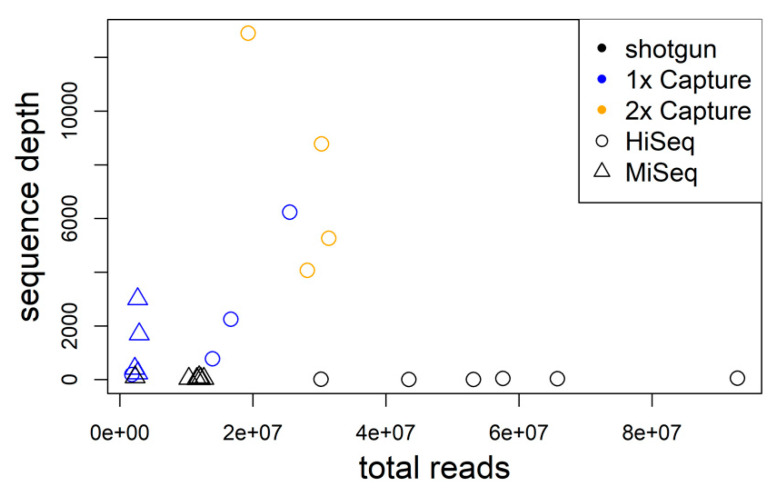
Research on the ecology and evolution of viruses is often hampered by the limitation of sequence information to short parts of the genomes or single genomes derived from cultures. In this study, we use hybrid sequence capture enrichment in combination with high-throughput sequencing to provide efficient access to full genomes of European hantaviruses from rodent samples obtained in the field. We applied this methodology to Tula (TULV) and Puumala (PUUV) orthohantaviruses for which analyses from natural host samples are typically restricted to partial sequences of their tri-segmented RNA genome. We assembled a total of ten novel hantavirus genomes with very high coverage (on average >99%) and sequencing depth (average >247×). A comparison with partial Sanger sequences indicated an accuracy of >99.9% for the assemblies. An analysis of two common vole () samples infected with two TULV strains each allowed for the assembly of all four TULV genomes. Combining the novel sequences with all available TULV and PUUV genomes revealed very similar patterns of sequence diversity along the genomes, except for remarkably higher diversity in the non-coding region of the S-segment in PUUV. The genomic distribution of polymorphisms in the coding sequence was similar between the species, but differed between the segments with the highest sequence divergence of 0.274 for the M-segment, 0.265 for the S-segment, and 0.248 for the L-segment (overall 0.258). Phylogenetic analyses showed the clustering of genome sequences consistent with their geographic distribution within each species. Genome-wide data yielded extremely high node support values, despite the impact of strong mutational saturation that is expected for hantavirus sequences obtained over large spatial distances. We conclude that genome sequencing based on capture enrichment protocols provides an efficient means for ecological and evolutionary investigations of hantaviruses at an unprecedented completeness and depth.
对病毒的生态学和进化的研究往往受到序列信息的限制,只能得到基因组的短片段或来自培养物的单个基因组。在这项研究中,我们使用混合序列捕获富集与高通量测序相结合,从野外获得的啮齿动物样本中有效地获得了欧洲汉坦病毒的全基因组。我们将这种方法应用于图尔病毒(TULV)和普马拉病毒(PUUV)正粘病毒,对于这些病毒,从其自然宿主样本中分析通常仅限于其三片段 RNA 基因组的部分序列。我们总共组装了十个新的汉坦病毒基因组,具有非常高的覆盖率(平均> 99%)和测序深度(平均> 247×)。与部分 Sanger 序列的比较表明,组装的准确性> 99.9%。对两个普通田鼠样本的分析表明,每个样本都感染了两种 TULV 株,从而可以组装所有四种 TULV 基因组。将新序列与所有可用的 TULV 和 PUUV 基因组相结合,揭示了沿基因组的序列多样性非常相似的模式,除了 PUUV 的 S 片段中非编码区的多样性明显更高。编码序列中的多态性在种间的基因组分布相似,但在片段间存在差异,其中 M 片段的序列差异最大,为 0.274,S 片段为 0.265,L 片段为 0.248(总体为 0.258)。系统发育分析表明,基因组序列的聚类与其在每个物种中的地理分布一致。尽管在获得大空间距离的汉坦病毒序列时预计会出现强烈的突变饱和,但全基因组数据产生了极高的节点支持值。我们得出的结论是,基于捕获富集方案的基因组测序为汉坦病毒的生态和进化研究提供了一种高效的方法,其完整性和深度前所未有。