Department of Biotechnology, Bio-Engineering and Drug Design Lab, Bhupat and Jyoti Mehta School of Biosciences, Indian Institute of Technology-Madras, Adayar, Chennai, Tamil Nadu, India.
Department of Biosciences, Sri Sathya Sai Institute of Higher Learning, Puttaparthi, Andhra Pradesh, India.
J Biomol Struct Dyn. 2021 Oct;39(16):6324-6337. doi: 10.1080/07391102.2020.1796802. Epub 2020 Jul 22.
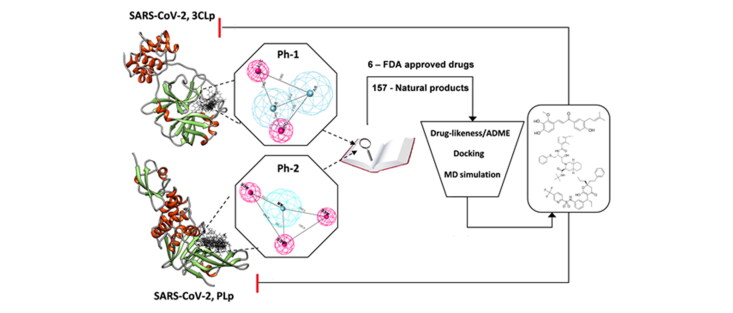
SARS-related coronaviruses poses continual threat to humanity by rapidly mutating and emerging as severe pandemic outbreaks, including the current nCoV-19 pandemic. Hence a rapid drug repositioning and lead identification strategy are required to mitigate these outbreaks. We report a pharmacophore and molecular dynamics-based approach for drug repositioning and lead identification against dual targets (3CLp and PLp) of SARS-CoV-2. The pharmacophore model of 3CLp inhibitors was apolar with two aromatic and two H-bond acceptors, whereas that of PLp was relatively polar, bearing one aromatic and three H-bond acceptors. Pharmacophore-based virtual screening yielded six existing FDA-approved drugs and twelve natural products with both the pharmacophoric features. Among them are nelfinavir, tipranavir and licochalcone-D, which has shown better binding characteristics with both the proteases compared to lopinavir. The molecular dynamics revealed that the connecting loop (residues 176-199) of 3CLp is highly flexible, and hence, inhibitors should avoid high-affinity interactions with it. Lopinavir, due to its high affinity with the loop region, exhibited unstable binding. Further, the van der Waals size of the 3CLp inhibitors positively correlated with their binding affinity with 3CLp. However, the van der Waals size of a ligand should not cross a threshold of 572Å, beyond which the ligands are likely to make high-affinity interaction with the loop and suffer unstable binding as observed in the case of lopinavir. Similarly, the total polar surface area of the ligands were found to be negatively correlated with their binding affinity with PLp.
SARS 相关冠状病毒通过快速突变并以严重的大流行爆发形式出现,包括当前的 nCoV-19 大流行,对人类构成持续威胁。因此,需要快速的药物重新定位和先导物识别策略来减轻这些爆发。我们报告了一种基于药效团和分子动力学的药物重新定位和先导物识别方法,针对 SARS-CoV-2 的两个靶标(3CLp 和 PLp)。3CLp 抑制剂的药效团模型具有非极性,包含两个芳基和两个氢键受体,而 PLp 的药效团模型则相对极性,具有一个芳基和三个氢键受体。基于药效团的虚拟筛选产生了六种现有的 FDA 批准的药物和十二种具有双重药效团特征的天然产物。其中包括奈非那韦、替普那韦和甘草查尔酮-D,它们与两种蛋白酶的结合特性均优于洛匹那韦。分子动力学揭示 3CLp 的连接环(残基 176-199)高度灵活,因此抑制剂应避免与它形成高亲和力相互作用。洛匹那韦由于与环区具有高亲和力,表现出不稳定的结合。此外,3CLp 抑制剂的范德华尺寸与它们与 3CLp 的结合亲和力呈正相关。然而,配体的范德华尺寸不应超过 572Å 的阈值,否则配体可能与环形成高亲和力相互作用,并像洛匹那韦那样表现出不稳定的结合。同样,配体的总极性表面积与它们与 PLp 的结合亲和力呈负相关。