Department of Pharmacognosy, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia.
Health and Basic Science Research Centre, Majmaah University, Majmaah 11952, Saudi Arabia; Department of Medical Laboratory Sciences, College of Applied Medical Sciences, Majmaah University, Majmaah 11952, Saudi Arabia.
J Infect Public Health. 2021 May;14(5):611-619. doi: 10.1016/j.jiph.2021.01.016. Epub 2021 Feb 9.
The emergence and spread of SARS-CoV-2 throughout the world has created an enormous socioeconomic impact. Although there are several promising drug candidates in clinical trials, none is available clinically. Thus, the drug repurposing approach may help to overcome the current pandemic.
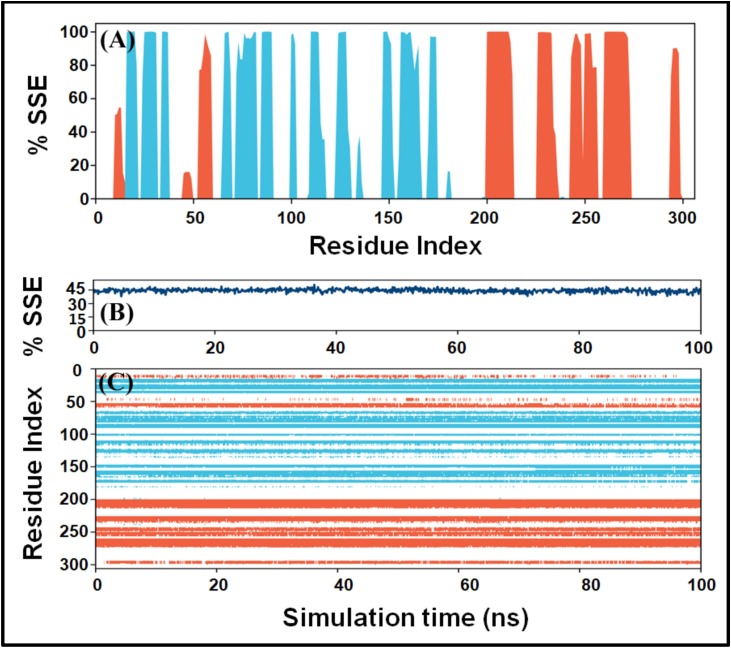
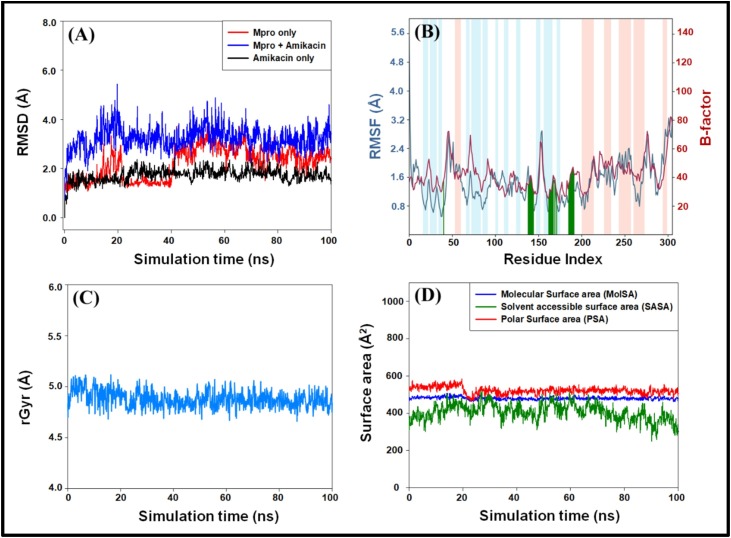
The main protease (M) of SARS-CoV-2 is crucial for cleaving nascent polypeptide chains. Here, FDA-approved antiviral and anti-infection drugs were screened by high-throughput virtual screening (HTVS) followed by re-docking with standard-precision (SP) and extra-precision (XP) molecular docking. The most potent drug's binding was further validated by free energy calculations (Prime/MM-GBSA) and molecular dynamics (MD) simulation.
Out of 1397 potential drugs, 157 showed considerable affinity toward M. After HTVS, SP, and XP molecular docking, four high-affinity lead drugs (Iodixanol, Amikacin, Troxerutin, and Rutin) with docking energies -10.629 to -11.776kcal/mol range were identified. Among them, Amikacin exhibited the lowest Prime/MM-GBSA energy (-73.800kcal/mol). It led us to evaluate other aminoglycosides (Neomycin, Paramomycin, Gentamycin, Streptomycin, and Tobramycin) against M. All aminoglycosides were bound to the substrate-binding site of M and interacted with crucial residues. Altogether, Amikacin was found to be the most potent inhibitor of M. MD simulations of the Amikacin-M complex suggested the formation of a complex stabilized by hydrogen bonds, salt bridges, and van der Waals interactions.
Aminoglycosides may serve as a scaffold to design potent drug molecules against COVID-19. However, further validation by in vitro and in vivo studies is required before using aminoglycosides as an anti-COVID-19 agent.
SARS-CoV-2 在全球范围内的出现和传播造成了巨大的社会经济影响。尽管临床试验中有几种有前途的候选药物,但没有一种可用于临床。因此,药物再利用方法可能有助于克服当前的大流行。
SARS-CoV-2 的主要蛋白酶(M)对于切割新生多肽链至关重要。在这里,通过高通量虚拟筛选(HTVS)筛选了 FDA 批准的抗病毒和抗感染药物,然后用标准精度(SP)和额外精度(XP)分子对接进行重新对接。通过自由能计算(Prime/MM-GBSA)和分子动力学(MD)模拟进一步验证了最有效药物的结合。
在 1397 种潜在药物中,有 157 种对 M 表现出相当大的亲和力。经过 HTVS、SP 和 XP 分子对接后,确定了四种具有高亲和力的先导药物(碘代醇、阿米卡星、曲克芦丁和芦丁),其对接能为-10.629 至-11.776kcal/mol。其中,阿米卡星的 Prime/MM-GBSA 能量最低(-73.800kcal/mol)。这促使我们评估了针对 M 的其他氨基糖苷类药物(新霉素、巴龙霉素、庆大霉素、链霉素和妥布霉素)。所有氨基糖苷类药物都与 M 的底物结合位点结合,并与关键残基相互作用。总的来说,阿米卡星是 M 最有效的抑制剂。阿米卡星-M 复合物的 MD 模拟表明,形成了一个由氢键、盐桥和范德华相互作用稳定的复合物。
氨基糖苷类药物可能成为设计针对 COVID-19 的有效药物分子的支架。然而,在将氨基糖苷类药物用作抗 COVID-19 药物之前,还需要通过体外和体内研究进行进一步验证。