Chierici Marco, Bussola Nicole, Marcolini Alessia, Francescatto Margherita, Zandonà Alessandro, Trastulla Lucia, Agostinelli Claudio, Jurman Giuseppe, Furlanello Cesare
Fondazione Bruno Kessler, Trento, Italy.
University of Trento, Trento, Italy.
Front Oncol. 2020 Jun 30;10:1065. doi: 10.3389/fonc.2020.01065. eCollection 2020.
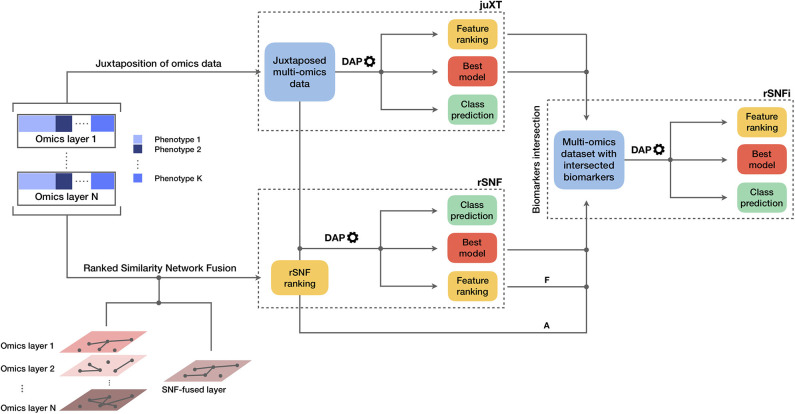
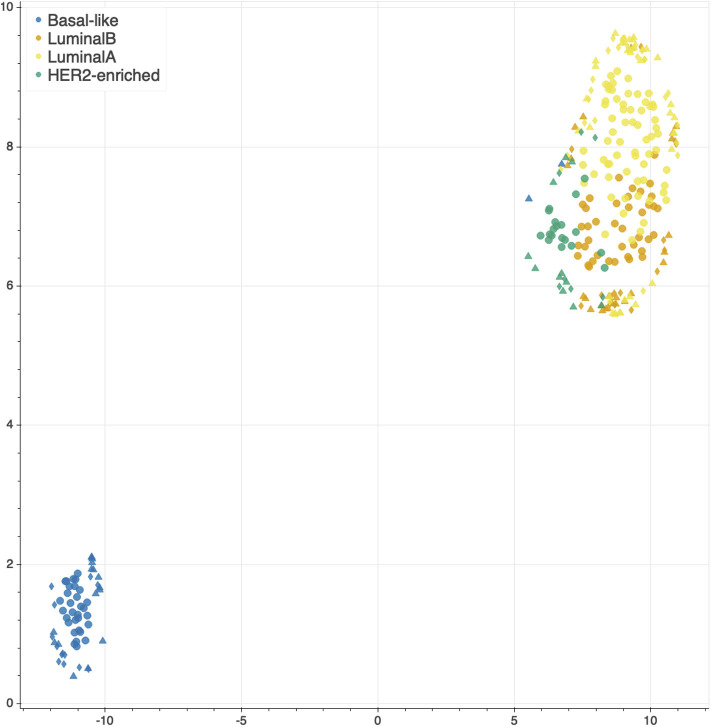
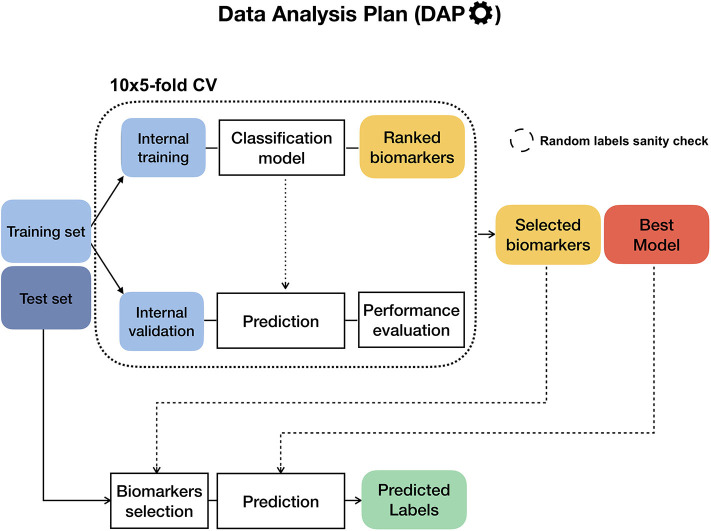
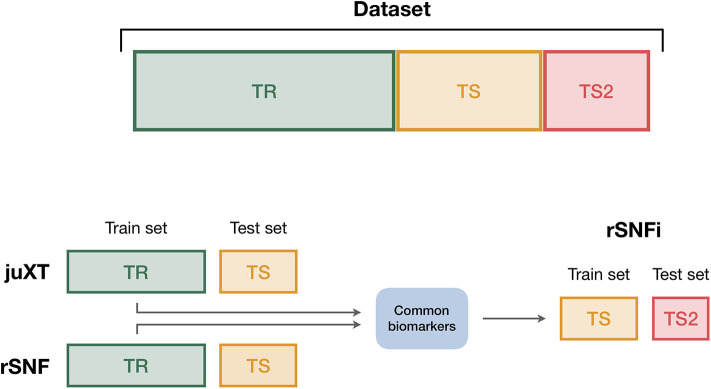
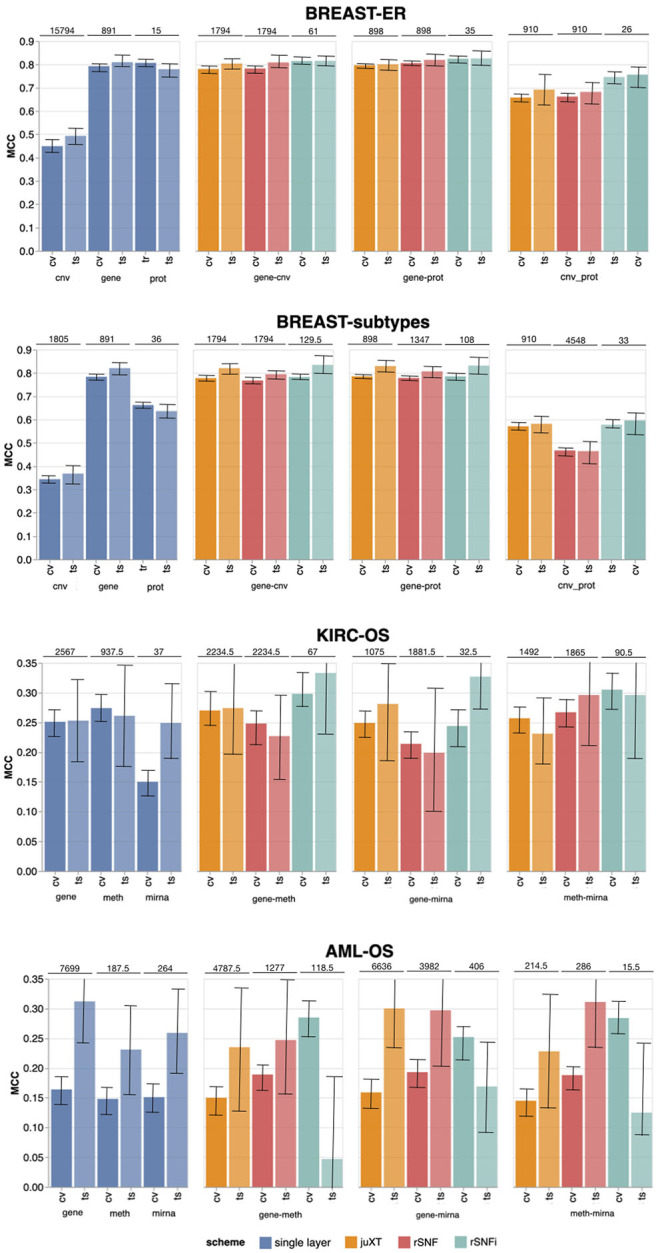
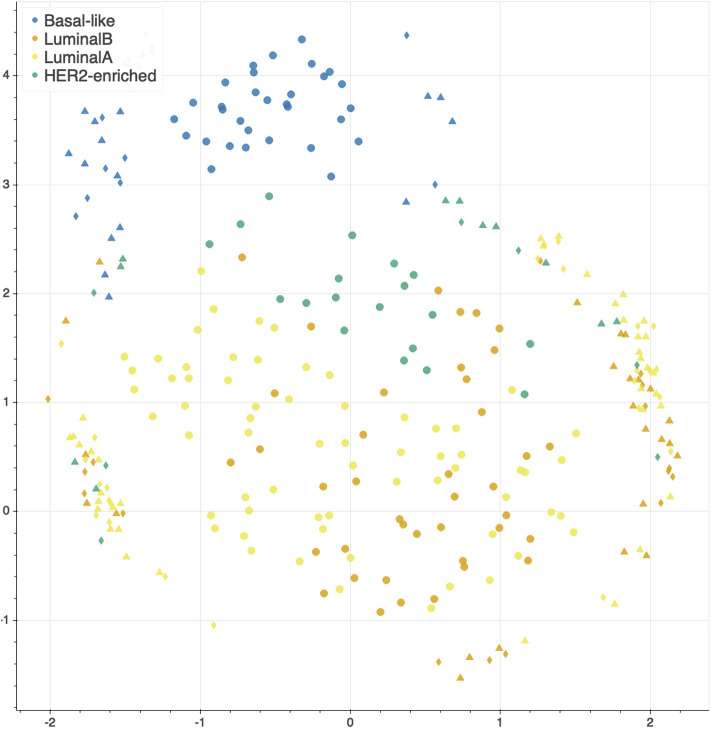
Recent technological advances and international efforts, such as The Cancer Genome Atlas (TCGA), have made available several pan-cancer datasets encompassing multiple omics layers with detailed clinical information in large collection of samples. The need has thus arisen for the development of computational methods aimed at improving cancer subtyping and biomarker identification from multi-modal data. Here we apply the Integrative Network Fusion (INF) pipeline, which combines multiple omics layers exploiting Similarity Network Fusion (SNF) within a machine learning predictive framework. INF includes a feature ranking scheme (rSNF) on SNF-integrated features, used by a classifier over juxtaposed multi-omics features (juXT). In particular, we show instances of INF implementing Random Forest (RF) and linear Support Vector Machine (LSVM) as the classifier, and two baseline RF and LSVM models are also trained on juXT. A compact RF model, called rSNFi, trained on the intersection of top-ranked biomarkers from the two approaches juXT and rSNF is finally derived. All the classifiers are run in a 10x5-fold cross-validation schema to warrant reproducibility, following the guidelines for an unbiased Data Analysis Plan by the US FDA-led initiatives MAQC/SEQC. INF is demonstrated on four classification tasks on three multi-modal TCGA oncogenomics datasets. Gene expression, protein expression and copy number variants are used to predict estrogen receptor status (BRCA-ER, = 381) and breast invasive carcinoma subtypes (BRCA-subtypes, = 305), while gene expression, miRNA expression and methylation data is used as predictor layers for acute myeloid leukemia and renal clear cell carcinoma survival (AML-OS, = 157; KIRC-OS, = 181). In test, INF achieved similar Matthews Correlation Coefficient (MCC) values and 97% to 83% smaller feature sizes (FS), compared with juXT for BRCA-ER (MCC: 0.83 vs. 0.80; FS: 56 vs. 1801) and BRCA-subtypes (0.84 vs. 0.80; 302 vs. 1801), improving KIRC-OS performance (0.38 vs. 0.31; 111 vs. 2319). INF predictions are generally more accurate in test than one-dimensional omics models, with smaller signatures too, where transcriptomics consistently play the leading role. Overall, the INF framework effectively integrates multiple data levels in oncogenomics classification tasks, improving over the performance of single layers alone and naive juxtaposition, and provides compact signature sizes.
近期的技术进步以及国际合作努力,如癌症基因组图谱(TCGA)项目,已提供了多个泛癌数据集,这些数据集包含多个组学层面的数据,并在大量样本中附有详细的临床信息。因此,开发旨在从多模态数据中改进癌症亚型分类和生物标志物识别的计算方法变得十分必要。在此,我们应用整合网络融合(INF)流程,该流程在机器学习预测框架内,利用相似性网络融合(SNF)整合多个组学层面的数据。INF包括一种基于SNF整合特征的特征排序方案(rSNF),由一个分类器用于并列的多组学特征(juXT)。具体而言,我们展示了将随机森林(RF)和线性支持向量机(LSVM)作为分类器的INF实例,并且还在juXT上训练了两个基线RF和LSVM模型。最终得出一个紧凑的RF模型,称为rSNFi,它是在juXT和rSNF这两种方法中排名靠前的生物标志物的交集上进行训练的。所有分类器均按照美国食品药品监督管理局(FDA)主导的MAQC/SEQC计划中无偏数据分析计划的指导方针,以10×5折交叉验证模式运行,以确保结果的可重复性。在三个多模态TCGA肿瘤基因组学数据集上的四项分类任务中对INF进行了验证。基因表达、蛋白质表达和拷贝数变异被用于预测雌激素受体状态(BRCA-ER,样本量 = 381)和乳腺浸润癌亚型(BRCA-亚型,样本量 = 305),而基因表达、miRNA表达和甲基化数据则被用作急性髓系白血病和肾透明细胞癌生存率(AML-OS,样本量 = 157;KIRC-OS,样本量 = 181)的预测层。在测试中,与juXT相比,INF在BRCA-ER(马修斯相关系数(MCC):0.83对0.80;特征数量(FS):56对1801)和BRCA-亚型(0.84对0.80;302对1801)上实现了相似的MCC值,且特征数量缩小了97%至83%,同时提高了KIRC-OS的性能(0.38对0.31;111对2319)。在测试中,INF的预测通常比一维组学模型更准确,特征标记也更小,其中转录组学始终发挥主导作用。总体而言,INF框架在肿瘤基因组学分类任务中有效地整合了多个数据层面,比单独的单层数据和简单并列的性能有所提升,并提供了紧凑的特征标记大小。