Department of Computer Science and Engineering, Aliah University, Kolkata, West Bengal, 700160, India.
Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, TX, 77030, USA.
BMC Bioinformatics. 2022 Apr 28;23(Suppl 3):153. doi: 10.1186/s12859-022-04678-y.
As many complex omics data have been generated during the last two decades, dimensionality reduction problem has been a challenging issue in better mining such data. The omics data typically consists of many features. Accordingly, many feature selection algorithms have been developed. The performance of those feature selection methods often varies by specific data, making the discovery and interpretation of results challenging.
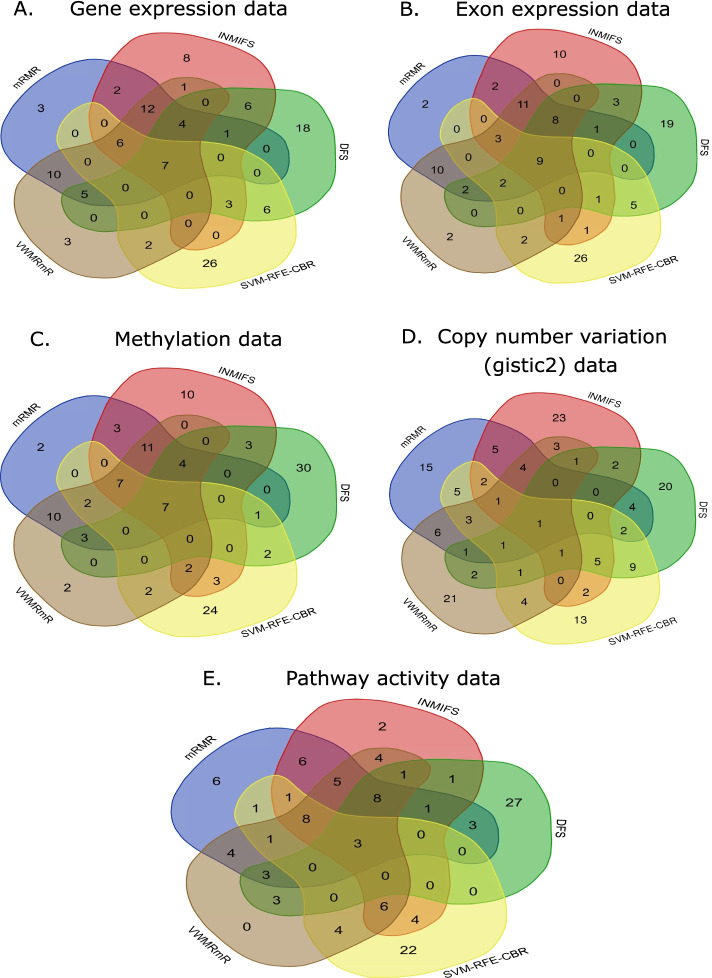
In this study, we performed a comprehensive comparative study of five widely used supervised feature selection methods (mRMR, INMIFS, DFS, SVM-RFE-CBR and VWMRmR) for multi-omics datasets. Specifically, we used five representative datasets: gene expression (Exp), exon expression (ExpExon), DNA methylation (hMethyl27), copy number variation (Gistic2), and pathway activity dataset (Paradigm IPLs) from a multi-omics study of acute myeloid leukemia (LAML) from The Cancer Genome Atlas (TCGA). The different feature subsets selected by the aforesaid five different feature selection algorithms are assessed using three evaluation criteria: (1) classification accuracy (Acc), (2) representation entropy (RE) and (3) redundancy rate (RR). Four different classifiers, viz., C4.5, NaiveBayes, KNN, and AdaBoost, were used to measure the classification accuary (Acc) for each selected feature subset. The VWMRmR algorithm obtains the best Acc for three datasets (ExpExon, hMethyl27 and Paradigm IPLs). The VWMRmR algorithm offers the best RR (obtained using normalized mutual information) for three datasets (Exp, Gistic2 and Paradigm IPLs), while it gives the best RR (obtained using Pearson correlation coefficient) for two datasets (Gistic2 and Paradigm IPLs). It also obtains the best RE for three datasets (Exp, Gistic2 and Paradigm IPLs). Overall, the VWMRmR algorithm yields best performance for all three evaluation criteria for majority of the datasets. In addition, we identified signature genes using supervised learning collected from the overlapped top feature set among five feature selection methods. We obtained a 7-gene signature (ZMIZ1, ENG, FGFR1, PAWR, KRT17, MPO and LAT2) for EXP, a 9-gene signature for ExpExon, a 7-gene signature for hMethyl27, one single-gene signature (PIK3CG) for Gistic2 and a 3-gene signature for Paradigm IPLs.
We performed a comprehensive comparison of the performance evaluation of five well-known feature selection methods for mining features from various high-dimensional datasets. We identified signature genes using supervised learning for the specific omic data for the disease. The study will help incorporate higher order dependencies among features.
在过去的二十年中,已经产生了许多复杂的组学数据,降维问题成为了更好地挖掘这些数据的一个具有挑战性的问题。组学数据通常由许多特征组成。因此,已经开发了许多特征选择算法。这些特征选择方法的性能通常因特定数据而异,这使得结果的发现和解释具有挑战性。
在这项研究中,我们对五种广泛使用的有监督特征选择方法(mRMR、INMIFS、DFS、SVM-RFE-CBR 和 VWMRmR)进行了全面的比较研究,用于多组学数据集。具体来说,我们使用了五个代表性数据集:来自癌症基因组图谱(TCGA)的急性髓系白血病(LAML)的多组学研究中的基因表达(Exp)、外显子表达(ExpExon)、DNA 甲基化(hMethyl27)、拷贝数变异(Gistic2)和途径活性数据集(Paradigm IPLs)。使用上述五种不同特征选择算法选择的不同特征子集使用三个评估标准进行评估:(1)分类准确率(Acc),(2)表示熵(RE)和(3)冗余率(RR)。使用四种不同的分类器,即 C4.5、朴素贝叶斯、KNN 和 AdaBoost,测量每个选定特征子集的分类准确率(Acc)。VWMRmR 算法在三个数据集(ExpExon、hMethyl27 和 Paradigm IPLs)中获得最佳 Acc。VWMRmR 算法在三个数据集(Exp、Gistic2 和 Paradigm IPLs)中提供最佳 RR(使用归一化互信息获得),而在两个数据集(Gistic2 和 Paradigm IPLs)中提供最佳 RR(使用皮尔逊相关系数获得)。它还在三个数据集(Exp、Gistic2 和 Paradigm IPLs)中获得最佳 RE。总体而言,VWMRmR 算法在大多数数据集的所有三个评估标准中均具有最佳性能。此外,我们使用来自五个特征选择方法中重叠的顶级特征集的监督学习来识别特征。我们获得了一个 7 个基因特征(ZMIZ1、ENG、FGFR1、PAWR、KRT17、MPO 和 LAT2)用于 EXP,一个 9 个基因特征用于 ExpExon,一个 7 个基因特征用于 hMethyl27,一个单基因特征(PIK3CG)用于 Gistic2,以及一个 3 个基因特征用于 Paradigm IPLs。
我们对五种知名特征选择方法的性能评估进行了全面比较,用于挖掘来自各种高维数据集的特征。我们使用监督学习为特定的组学数据识别疾病的特征基因。该研究将有助于整合特征之间的更高阶相关性。