Hubrecht Institute, Royal Netherlands Academy of Arts and Sciences and University Medical Center Utrecht, 3584 CT Utrecht, The Netherlands.
Department of Cardiology, University Medical Centre Utrecht, Utrecht, The Netherlands.
Cardiovasc Res. 2021 May 25;117(6):1532-1545. doi: 10.1093/cvr/cvaa233.
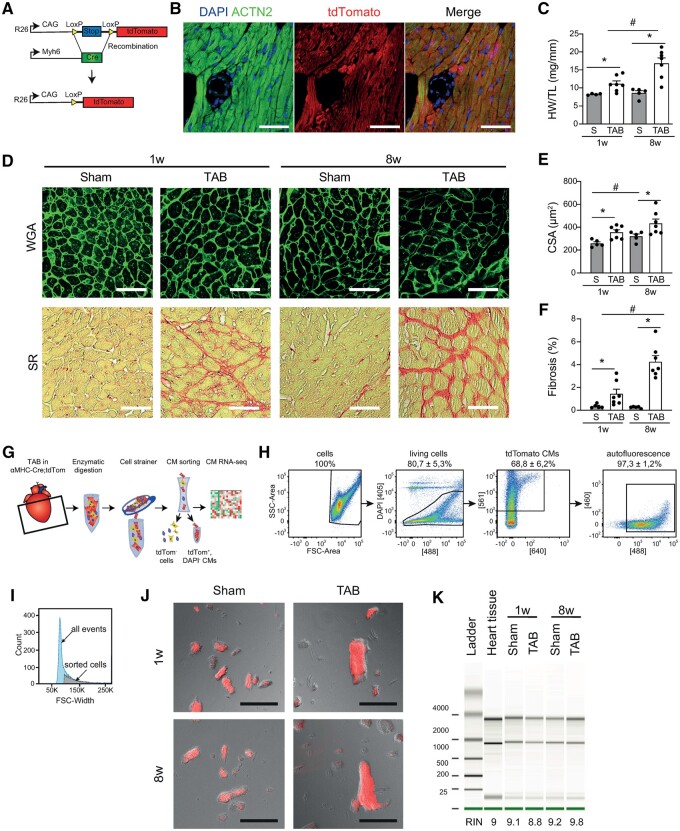
Pathological cardiac remodelling is characterized by cardiomyocyte (CM) hypertrophy and fibroblast activation, which can ultimately lead to maladaptive hypertrophy and heart failure (HF). Genome-wide expression analysis on heart tissue has been instrumental for the identification of molecular mechanisms at play. However, these data were based on signals derived from all cardiac cell types. Here, we aimed for a more detailed view on molecular changes driving maladaptive CM hypertrophy to aid in the development of therapies to reverse pathological remodelling.
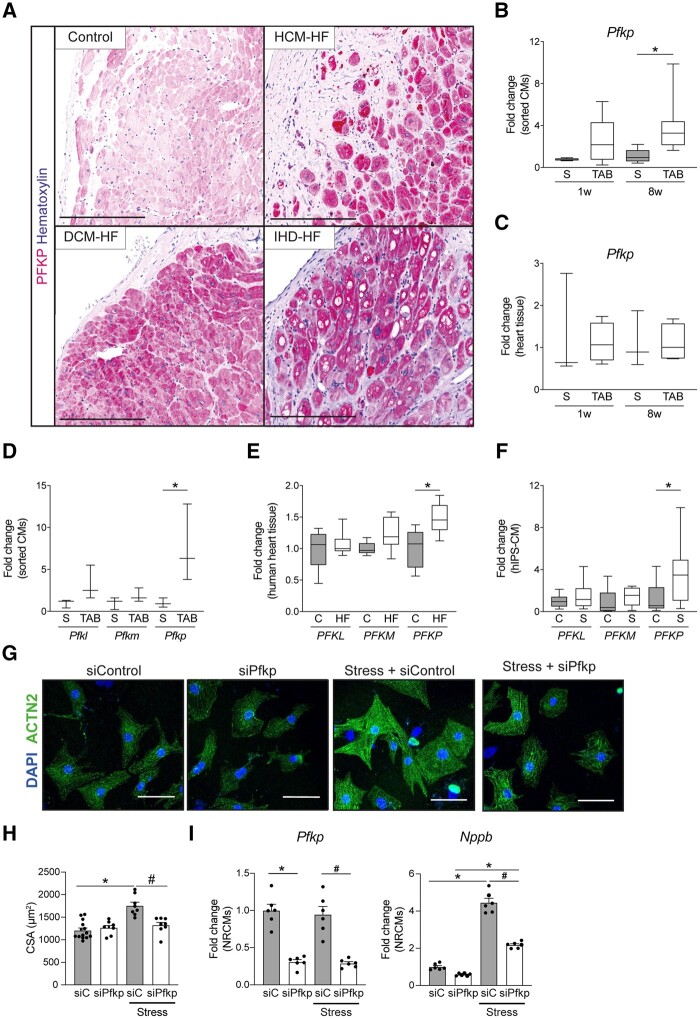
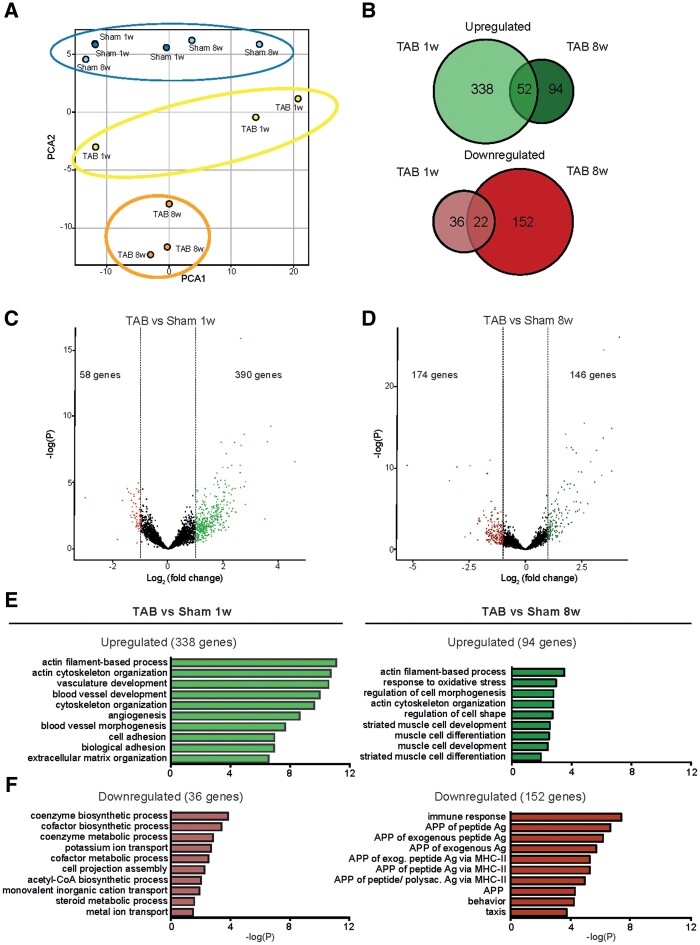
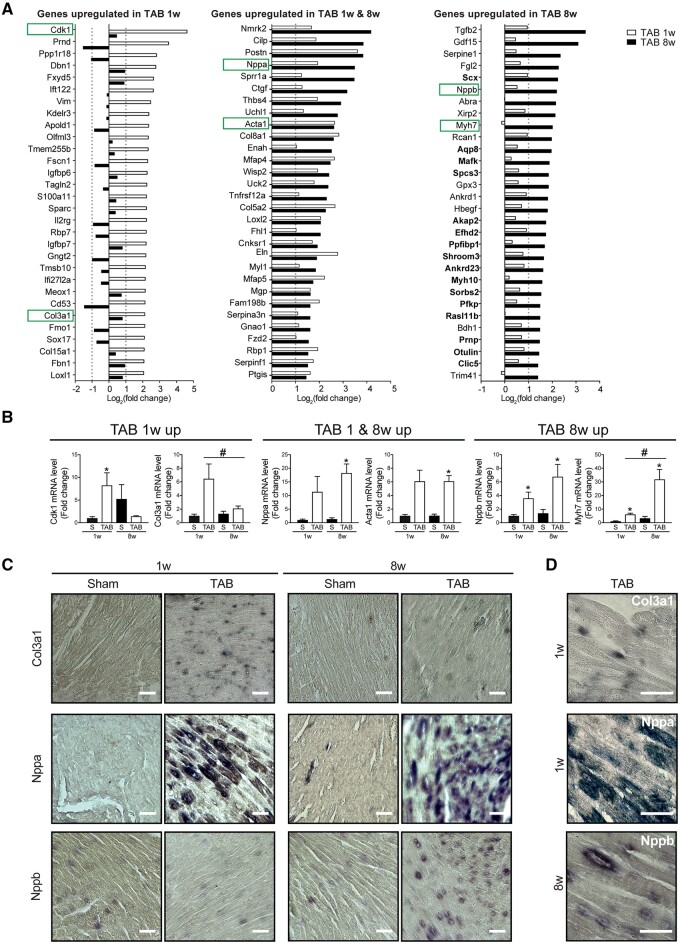
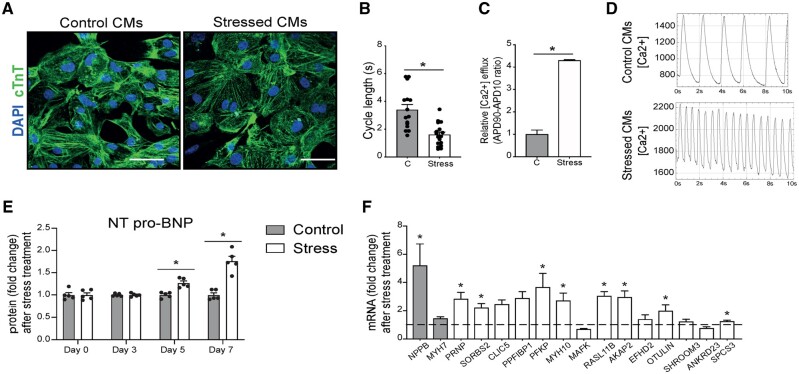
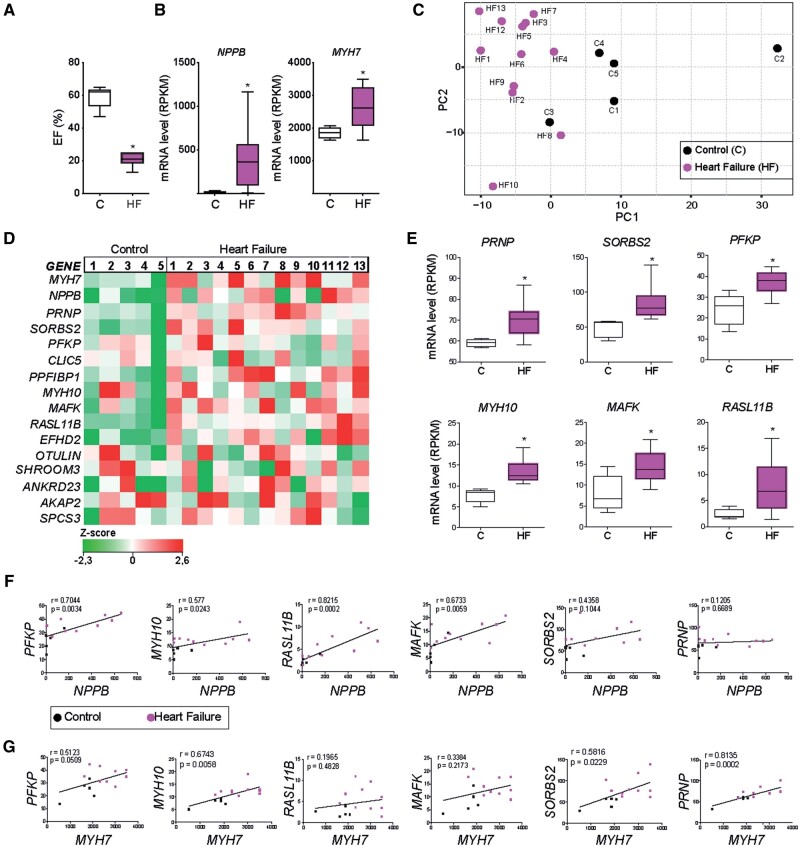
Utilizing CM-specific reporter mice exposed to pressure overload by transverse aortic banding and CM isolation by flow cytometry, we obtained gene expression profiles of hypertrophic CMs in the more immediate phase after stress, and CMs showing pathological hypertrophy. We identified subsets of genes differentially regulated and specific for either stage. Among the genes specifically up-regulated in the CMs during the maladaptive phase we found known stress markers, such as Nppb and Myh7, but additionally identified a set of genes with unknown roles in pathological hypertrophy, including the platelet isoform of phosphofructokinase (PFKP). Norepinephrine-angiotensin II treatment of cultured human CMs induced the secretion of N-terminal-pro-B-type natriuretic peptide (NT-pro-BNP) and recapitulated the up-regulation of these genes, indicating conservation of the up-regulation in failing CMs. Moreover, several genes induced during pathological hypertrophy were also found to be increased in human HF, with their expression positively correlating to the known stress markers NPPB and MYH7. Mechanistically, suppression of Pfkp in primary CMs attenuated stress-induced gene expression and hypertrophy, indicating that Pfkp is an important novel player in pathological remodelling of CMs.
Using CM-specific transcriptomic analysis, we identified novel genes induced during pathological hypertrophy that are relevant for human HF, and we show that PFKP is a conserved failure-induced gene that can modulate the CM stress response.
病理性心脏重构的特征是心肌细胞(CM)肥大和成纤维细胞激活,最终导致适应性不良的肥大和心力衰竭(HF)。对心脏组织进行全基因组表达分析对于鉴定发挥作用的分子机制至关重要。然而,这些数据基于来自所有心肌细胞类型的信号。在这里,我们旨在更详细地观察驱动适应性不良 CM 肥大的分子变化,以帮助开发逆转病理性重构的治疗方法。
利用 CM 特异性报告小鼠通过横主动脉缩窄暴露于压力超负荷,并通过流式细胞术分离 CM,我们获得了应激后更直接阶段的肥大 CM 的基因表达谱,以及表现出病理性肥大的 CM。我们鉴定了差异调节和特定于任一阶段的基因子集。在适应性不良阶段 CM 中特异性上调的基因中,我们发现了已知的应激标志物,如 Nppb 和 Myh7,但另外还发现了一组在病理性肥大中具有未知作用的基因,包括磷酸果糖激酶(PFKP)的血小板同工型。培养的人 CM 中去甲肾上腺素-血管紧张素 II 处理诱导 N 端前 B 型利钠肽(NT-pro-BNP)的分泌,并重新激活这些基因的上调,表明衰竭 CM 中的上调保持不变。此外,在病理性肥大期间诱导的几个基因也在人类 HF 中增加,其表达与已知的应激标志物 NPPB 和 MYH7 呈正相关。在机制上,在原代 CM 中抑制 Pfkp 可减弱应激诱导的基因表达和肥大,表明 Pfkp 是 CM 病理性重构的一个重要新的调节因子。
使用 CM 特异性转录组分析,我们鉴定了在病理性肥大期间诱导的与人类 HF 相关的新基因,并且我们表明 PFKP 是一个保守的衰竭诱导基因,可以调节 CM 的应激反应。