Liu Xiaoxuan, Duan Xiaohui, Zhang Yingshuang, Fan Dongsheng
Department of Neurology, Peking University Third Hospital, Beijing, China.
Department of Neurology, China-Japan Friendship Hospital, Beijing, China.
Front Neurol. 2020 Jul 3;11:630. doi: 10.3389/fneur.2020.00630. eCollection 2020.
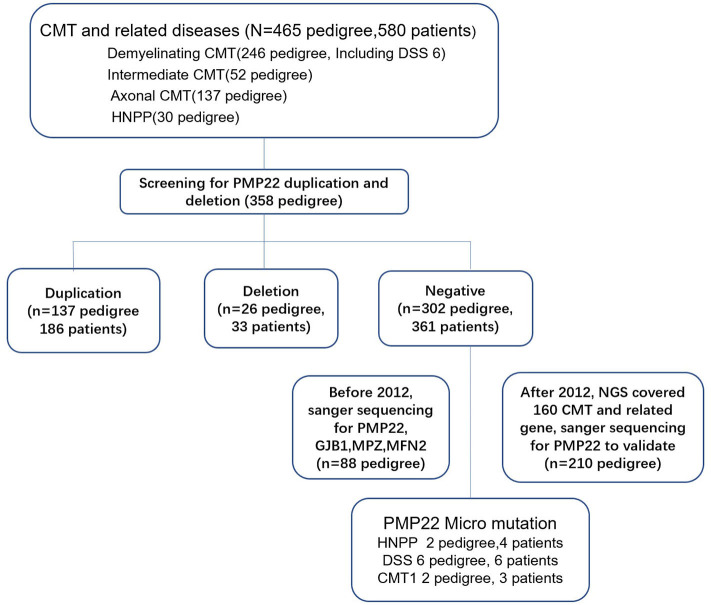
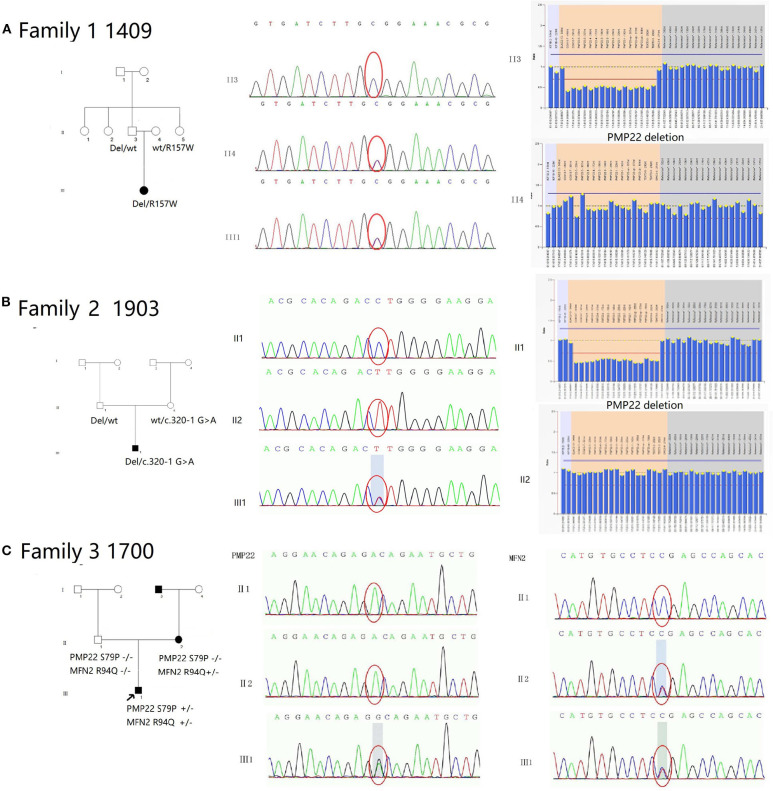
Charcot-Marie-Tooth (CMT) disease is a clinically and genetically heterogeneous group of inherited neuropathies. The purpose of this study is to identify the clinical and genetic diversity of peripheral myelin protein 22 () in Chinese patients with CMT disease and evaluate their correlations with the clinical manifestations. Using the multiplex ligation-dependent probe amplification (MLPA) technique and Sanger sequencing of in a cohort of 465 Chinese families between 2007 and 2019, we identified 137 pedigrees with duplications (29.5%), 26 pedigrees with deletions (5.6%), and 10 pedigrees with point mutations (2.2%). By comparing our data with the results from other CMT centers in China, we estimate that the frequency of mutation in mainland China is ~23.3% (261/1120). We confirmed mutations in 40% (4/10) of point mutations. We have also identified two severely affected patients who are compound heterozygotes for recessive mutations (novel mutation c.320-1 G>A and R157W mutation) and a 1.5 Mb deletion in 17p11.2-p12, suggesting that c.320-1 G>A might be another recessive allele contributing to DSS in addition to the T118M and R157W mutations. A mutation of S79P in was also identified concomitantly with the R94W mutation in mitofusin2 (). Our study highlights the phenotypic variability associated with mutations in mainland China. The results provide valuable insights into the current strategy of genetic testing for CMT disease. NGS technology has increased the potential for efficient detection of variants of unknown significance (VUS) and concurrent causative genes. Greater cooperation between neurologists and molecular biologists is needed in future investigations.
夏科-马里-图思(CMT)病是一组临床和遗传异质性的遗传性神经病变。本研究的目的是确定中国CMT病患者外周髓鞘蛋白22(PMP22)的临床和遗传多样性,并评估它们与临床表现的相关性。在2007年至2019年间,我们对465个中国家庭组成的队列使用多重连接依赖探针扩增(MLPA)技术和PMP22的桑格测序,我们鉴定出137个家系存在PMP22重复(29.5%),26个家系存在PMP22缺失(5.6%),10个家系存在点突变(2.2%)。通过将我们的数据与中国其他CMT中心的结果进行比较,我们估计中国大陆PMP22突变的频率约为23.3%(261/1120)。我们在40%(4/10)的PMP22点突变中确认了突变。我们还鉴定出两名严重受累患者,他们是隐性PMP22突变(新突变c.320-1 G>A和R157W突变)的复合杂合子,以及17p11.2-p12区域1.5 Mb的缺失,这表明除了T118M和R157W突变外,c.320-1 G>A可能是导致严重脱髓鞘性感觉神经病(DSS)的另一个隐性等位基因。还同时鉴定出PMP22的S79P突变与线粒体融合蛋白2(MFN2)的R94W突变。我们的研究突出了中国大陆与PMP22突变相关的表型变异性。这些结果为当前CMT病的基因检测策略提供了有价值的见解。二代测序(NGS)技术增加了有效检测意义未明变异(VUS)和并发致病基因的潜力。未来的研究需要神经科医生和分子生物学家之间加强合作。