Department of Biological Sciences, University of Arkansas, Fayetteville, AR, 72701, USA.
Present address: Molecular Ecology Laboratory, Southwestern Native Aquatic Resources and Recovery Center (SNARRC), U.S. Fish & Wildlife Service, PO Box 219, Dexter, NM, 88230, USA.
BMC Bioinformatics. 2020 Jul 29;21(1):337. doi: 10.1186/s12859-020-03701-4.
Research on the molecular ecology of non-model organisms, while previously constrained, has now been greatly facilitated by the advent of reduced-representation sequencing protocols. However, tools that allow these large datasets to be efficiently parsed are often lacking, or if indeed available, then limited by the necessity of a comparable reference genome as an adjunct. This, of course, can be difficult when working with non-model organisms. Fortunately, pipelines are currently available that avoid this prerequisite, thus allowing data to be a priori parsed. An oft-used molecular ecology program (i.e., STRUCTURE), for example, is facilitated by such pipelines, yet they are surprisingly absent for a second program that is similarly popular and computationally more efficient (i.e., ADMIXTURE). The two programs differ in that ADMIXTURE employs a maximum-likelihood framework whereas STRUCTURE uses a Bayesian approach, yet both produce similar results. Given these issues, there is an overriding (and recognized) need among researchers in molecular ecology for bioinformatic software that will not only condense output from replicated ADMIXTURE runs, but also infer from these data the optimal number of population clusters (K).
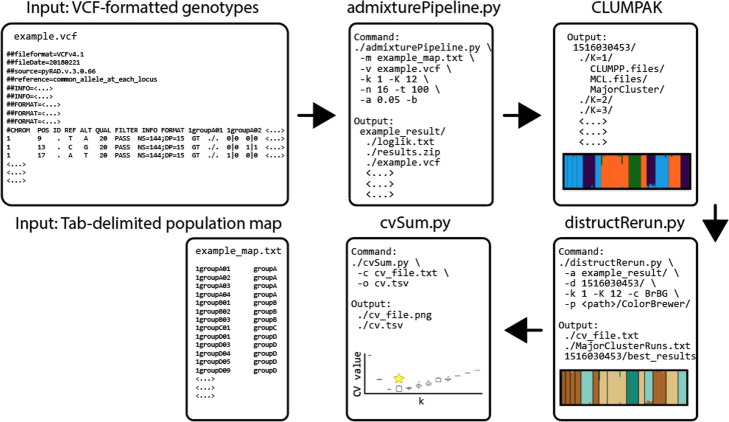
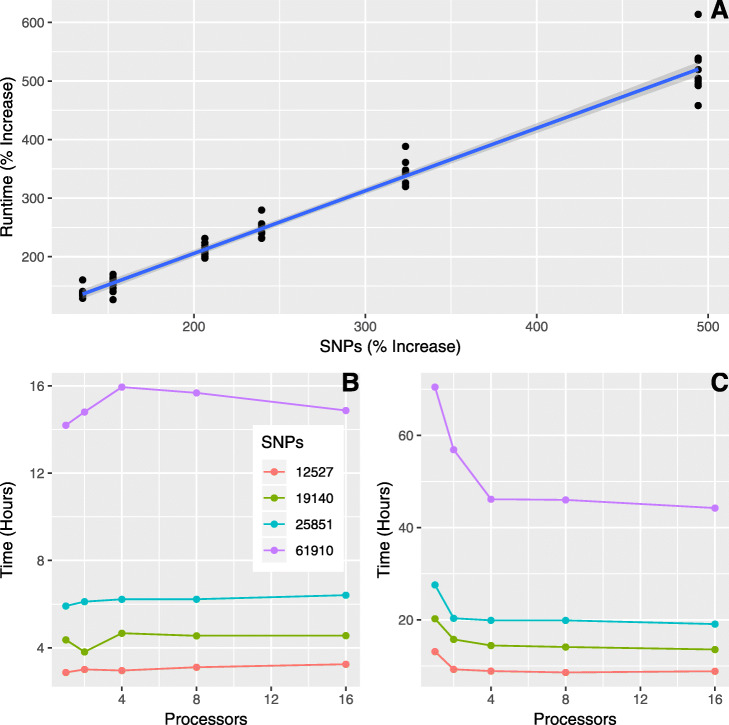
Here we provide such a program (i.e., ADMIXPIPE) that (a) filters SNPs to allow the delineation of population structure in ADMIXTURE, then (b) parses the output for summarization and graphical representation via CLUMPAK. Our benchmarks effectively demonstrate how efficient the pipeline is for processing large, non-model datasets generated via double digest restriction-site associated DNA sequencing (ddRAD). Outputs not only parallel those from STRUCTURE, but also visualize the variation among individual ADMIXTURE runs, so as to facilitate selection of the most appropriate K-value.
ADMIXPIPE successfully integrates ADMIXTURE analysis with popular variant call format (VCF) filtering software to yield file types readily analyzed by CLUMPAK. Large population genomic datasets derived from non-model organisms are efficiently analyzed via the parallel-processing capabilities of ADMIXTURE. ADMIXPIPE is distributed under the GNU Public License and freely available for Mac OSX and Linux platforms at: https://github.com/stevemussmann/admixturePipeline .
非模式生物的分子生态学研究虽然以前受到限制,但现在随着简化代表性测序协议的出现,已经得到了极大的促进。然而,允许有效解析这些大型数据集的工具往往缺乏,或者即使确实存在,也受到需要类似参考基因组作为辅助的限制。当然,在处理非模式生物时,这可能很困难。幸运的是,目前有一些管道可以避免这个前提条件,从而允许数据进行先验解析。例如,一个常用的分子生态学程序(即 STRUCTURE)就可以通过这些管道来实现,但是对于另一个同样受欢迎且计算效率更高的程序(即 ADMIXTURE),却没有类似的工具。这两个程序的区别在于 ADMIXTURE 采用最大似然框架,而 STRUCTURE 采用贝叶斯方法,但它们都产生类似的结果。鉴于这些问题,分子生态学研究人员迫切需要一种生物信息学软件,不仅可以压缩 ADMIXTURE 重复运行的输出,还可以从这些数据中推断出最佳的群体聚类数(K)。
在这里,我们提供了这样一个程序(即 ADMIXPIPE),它(a)过滤 SNP 以允许在 ADMIXTURE 中描绘群体结构,然后(b)通过 CLUMPAK 解析输出以进行总结和图形表示。我们的基准测试有效地证明了该管道处理通过双消化限制位点相关 DNA 测序(ddRAD)生成的大型非模式数据集的效率。输出不仅与 STRUCTURE 的输出平行,还可视化了个体 ADMIXTURE 运行之间的差异,从而便于选择最合适的 K 值。
ADMIXPIPE 成功地将 ADMIXTURE 分析与流行的变体调用格式(VCF)过滤软件集成在一起,生成易于通过 CLUMPAK 分析的文件类型。通过 ADMIXTURE 的并行处理能力,高效地分析了来自非模式生物的大型群体基因组数据集。ADMIXPIPE 是根据 GNU 公共许可证分发的,可以在 Mac OSX 和 Linux 平台上从以下网址免费获得:https://github.com/stevemussmann/admixturePipeline 。