Institute of Genetic Epidemiology, Medical Center-University of Freiburg, Freiburg, Germany
Institute of Genetic Epidemiology, Medical Center-University of Freiburg, Freiburg, Germany.
J Am Soc Nephrol. 2020 Oct;31(10):2326-2340. doi: 10.1681/ASN.2020010051. Epub 2020 Aug 6.
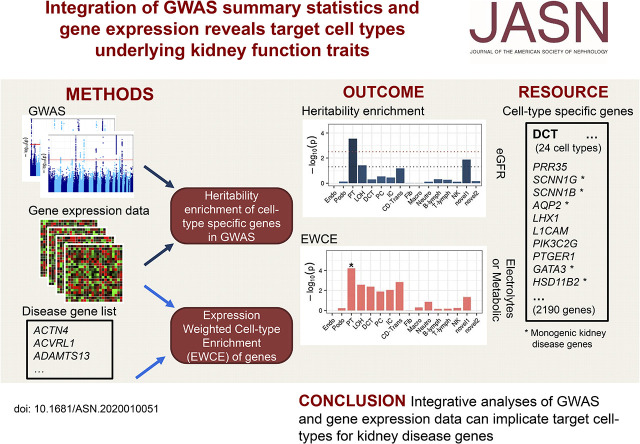
Genetic variants identified in genome-wide association studies (GWAS) are often not specific enough to reveal complex underlying physiology. By integrating RNA-seq data and GWAS summary statistics, novel computational methods allow unbiased identification of trait-relevant tissues and cell types.
The CKDGen consortium provided GWAS summary data for eGFR, urinary albumin-creatinine ratio (UACR), BUN, and serum urate. Genotype-Tissue Expression Project (GTEx) RNA-seq data were used to construct the top 10% specifically expressed genes for each of 53 tissues followed by linkage disequilibrium (LD) score-based enrichment testing for each trait. Similar procedures were performed for five kidney single-cell RNA-seq datasets from humans and mice and for a microdissected tubule RNA-seq dataset from rat. Gene set enrichment analyses were also conducted for genes implicated in Mendelian kidney diseases.
Across 53 tissues, genes in kidney function-associated GWAS loci were enriched in kidney (=9.1E-8 for eGFR; =1.2E-5 for urate) and liver (=6.8·10 for eGFR). In the kidney, proximal tubule was enriched in humans (=8.5E-5 for eGFR; =7.8E-6 for urate) and mice (=0.0003 for eGFR; =0.0002 for urate) and confirmed as the primary cell type in microdissected tubules and organoids. Gene set enrichment analysis supported this and showed enrichment of genes implicated in monogenic glomerular diseases in podocytes. A systematic approach generated a comprehensive list of GWAS genes prioritized by cell type-specific expression.
Integration of GWAS statistics of kidney function traits and gene expression data identified relevant tissues and cell types, as a basis for further mechanistic studies to understand GWAS loci.
全基因组关联研究(GWAS)中鉴定的遗传变异通常不够具体,无法揭示复杂的潜在生理机制。通过整合 RNA-seq 数据和 GWAS 汇总统计数据,新的计算方法可以在不偏倚的情况下识别与性状相关的组织和细胞类型。
CKDGen 联盟提供了 eGFR、尿白蛋白肌酐比(UACR)、BUN 和血清尿酸的 GWAS 汇总数据。使用基因型组织表达项目(GTEx)RNA-seq 数据为 53 种组织中的每一种构建了前 10%特异性表达的基因,然后对每种性状进行基于连锁不平衡(LD)得分的富集测试。对来自人类和小鼠的五个肾脏单细胞 RNA-seq 数据集以及大鼠微分离管 RNA-seq 数据集进行了类似的处理。还对孟德尔肾脏疾病相关基因进行了基因集富集分析。
在 53 种组织中,与肾脏功能相关的 GWAS 基因座中的基因在肾脏中富集(eGFR 为 9.1E-8;尿酸为 1.2E-5)和肝脏(eGFR 为 6.8·10)。在肾脏中,近端小管在人类中富集(eGFR 为 8.5E-5;尿酸为 7.8E-6)和小鼠(eGFR 为 0.0003;尿酸为 0.0002),并在微分离管和类器官中被确认为主要的细胞类型。基因集富集分析支持这一点,并显示出与单基因肾小球疾病相关基因在足细胞中的富集。系统的方法生成了一个由细胞类型特异性表达优先排序的 GWAS 基因的综合列表。
整合肾脏功能性状的 GWAS 统计数据和基因表达数据,确定了相关的组织和细胞类型,为进一步的机制研究提供了基础,以了解 GWAS 基因座。