Berbers Bas, Ceyssens Pieter-Jan, Bogaerts Pierre, Vanneste Kevin, Roosens Nancy H C, Marchal Kathleen, De Keersmaecker Sigrid C J
Transversal Activities in Applied Genomics, Sciensano, 1050 Brussels, Belgium.
Department of Information Technology, IDLab, Ghent University, IMEC, 9052 Ghent, Belgium.
Antibiotics (Basel). 2020 Aug 11;9(8):503. doi: 10.3390/antibiotics9080503.
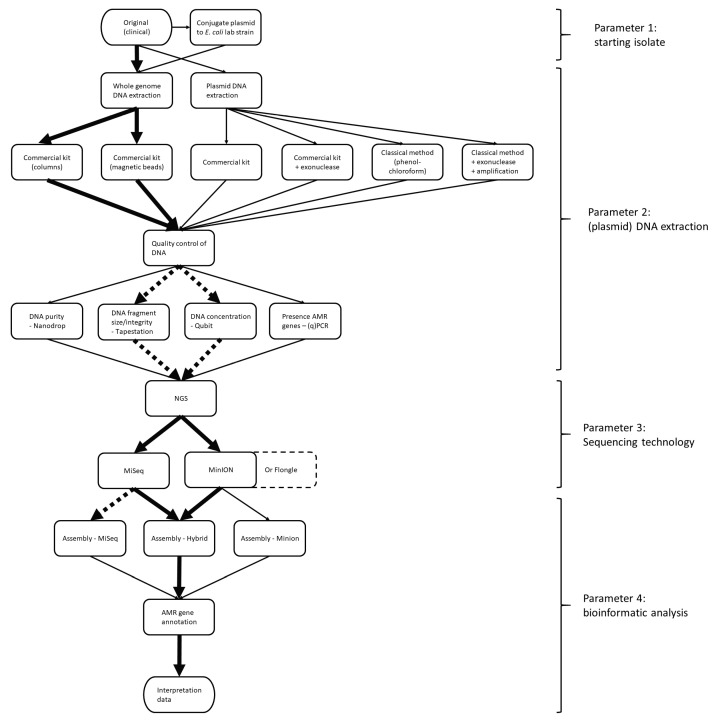
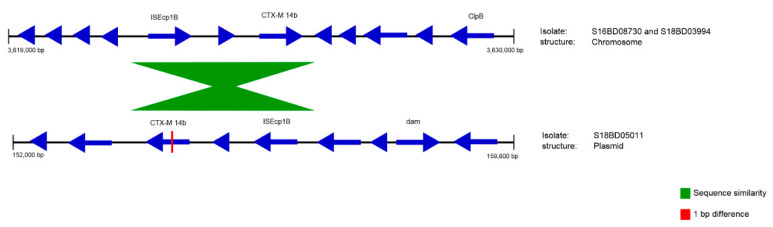
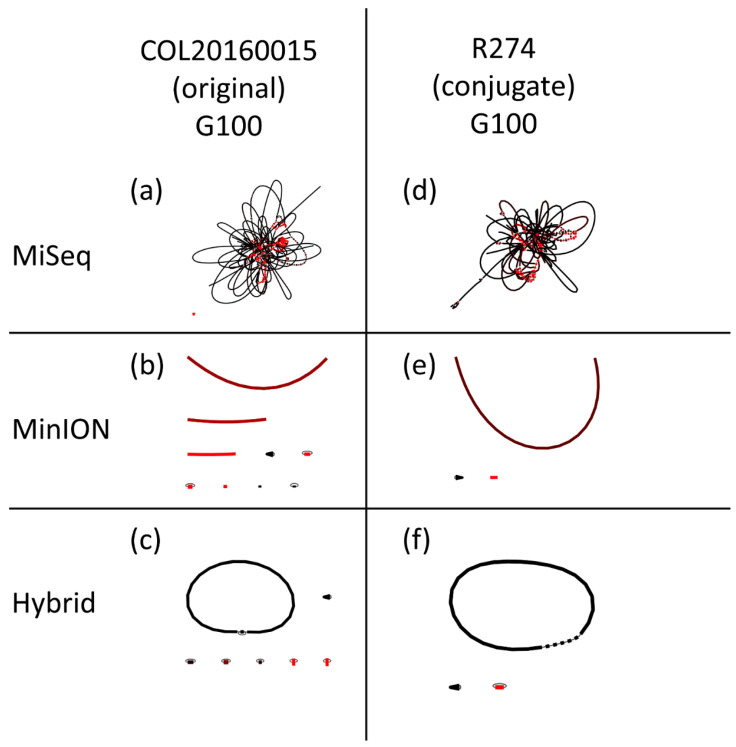
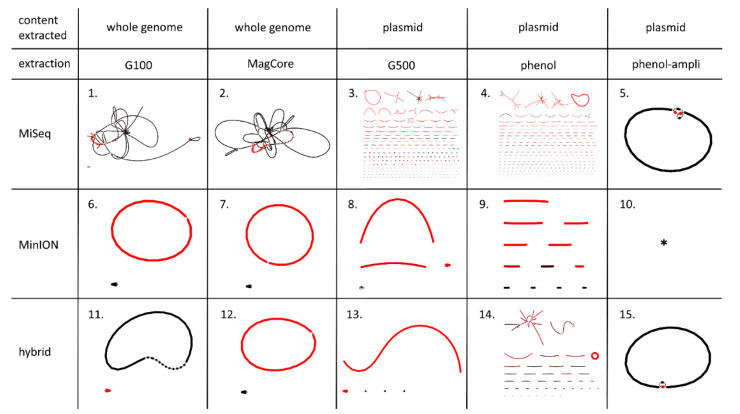
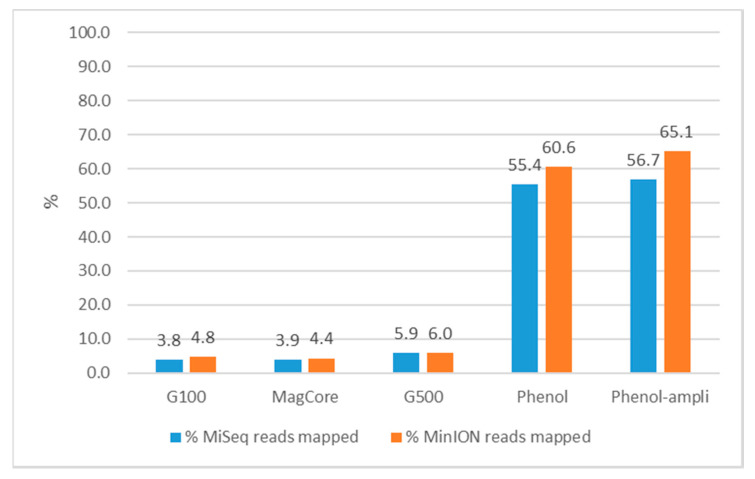
Antimicrobial resistance (AMR) is one of the most prominent public health threats. AMR genes localized on plasmids can be easily transferred between bacterial isolates by horizontal gene transfer, thereby contributing to the spread of AMR. Next-generation sequencing (NGS) technologies are ideal for the detection of AMR genes; however, reliable reconstruction of plasmids is still a challenge due to large repetitive regions. This study proposes a workflow to reconstruct plasmids with NGS data in view of AMR gene localization, i.e., chromosomal or on a plasmid. Whole-genome and plasmid DNA extraction methods were compared, as were assemblies consisting of short reads (Illumina MiSeq), long reads (Oxford Nanopore Technologies) and a combination of both (hybrid). Furthermore, the added value of conjugation of a plasmid to a known host was evaluated. As a case study, an isolate harboring a large, low-copy -carrying plasmid (>200 kb) was used. Hybrid assemblies of NGS data obtained from whole-genome DNA extractions of the original isolates resulted in the most complete reconstruction of plasmids. The optimal workflow was successfully applied to multidrug-resistant Kentucky isolates, where the transfer of an ESBL-gene-containing fragment from a plasmid to the chromosome was detected. This study highlights a strategy including wet and dry lab parameters that allows accurate plasmid reconstruction, which will contribute to an improved monitoring of circulating plasmids and the assessment of their risk of transfer.
抗菌药物耐药性(AMR)是最突出的公共卫生威胁之一。位于质粒上的AMR基因可通过水平基因转移在细菌分离株之间轻松转移,从而促进AMR的传播。下一代测序(NGS)技术是检测AMR基因的理想方法;然而,由于存在大量重复区域,可靠地重建质粒仍然是一项挑战。本研究提出了一种根据AMR基因定位(即位于染色体上或质粒上)利用NGS数据重建质粒的工作流程。比较了全基因组和质粒DNA提取方法,以及由短读长(Illumina MiSeq)、长读长(Oxford Nanopore Technologies)和两者结合(混合)组成的组装方法。此外,还评估了将质粒与已知宿主进行接合的附加价值。作为案例研究,使用了一株携带大型低拷贝质粒(>200 kb)的分离株。从原始分离株的全基因组DNA提取中获得的NGS数据的混合组装导致了质粒的最完整重建。该优化工作流程已成功应用于多重耐药的肯塔基分离株,检测到了含ESBL基因的片段从质粒转移到染色体上。本研究突出了一种包括湿实验室和干实验室参数的策略,该策略可实现准确的质粒重建,这将有助于改进对循环质粒的监测及其转移风险评估。