Bioinformatics Research Laboratory, Dr. D. Y. Patil Biotechnology and Bioinformatics Institute, Dr. D. Patil Vidyapeeth, Pune, India.
Bioinformatics Research Group, MIT School of Bioengineering Science and Research, MIT- ADT University, Pune, India.
J Biomol Struct Dyn. 2021 Nov;39(18):7294-7305. doi: 10.1080/07391102.2020.1805019. Epub 2020 Aug 20.
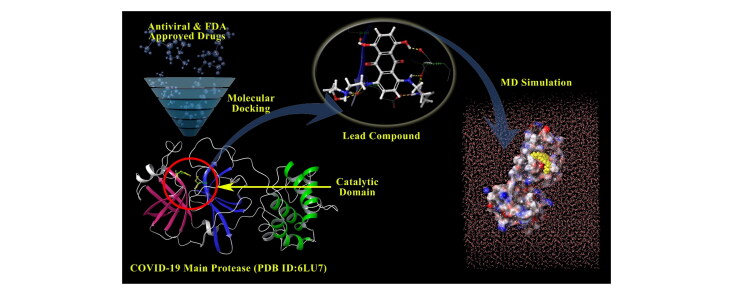
The outbreak of novel coronavirus (COVID-19), which began from Wuhan City, Hubei, China, and declared as a Public Health Emergency of International Concern by World Health Organization (WHO) on 30 January 2020. The present study describes how the available drug candidates can be used as a potential SARS-CoV-2 M inhibitor by molecular docking and molecular dynamic simulation studies. Drug repurposing strategy is applied by using the library of antiviral and FDA approved drugs retrieved from the Selleckchem Inc. (Houston, TX, http://www.selleckchem.com) and DrugBank database respectively. Computational methods like molecular docking and molecular dynamics simulation were used. The molecular docking calculations were performed using LeadIT FlexX software. The molecular dynamics simulations of 100 ns were performed to study conformational stability for all complex systems. Mitoxantrone and Leucovorin from FDA approved drug library and Birinapant and Dynasore from anti-viral drug libraries interact with SARS-CoV-2 M at higher efficiency as a result of the improved steric and hydrophobic environment in the binding cavity to make stable complex. Also, the molecular dynamics simulations of 100 ns revealed the mean RMSD value of 2.25 Å for all the complex systems. This shows that lead compounds bound tightly within the M cavity and thus having conformational stability. Glutamic acid (Glu166) of M is a key residue to hold and form a stable complex of reported lead compounds by forming hydrogen bonds and salt bridge. Our findings suggest that Mitoxantrone, Leucovorin, Birinapant, and Dynasore represents potential inhibitors of SARS-CoV-2 M.
新型冠状病毒(COVID-19)爆发于中国湖北省武汉市,世界卫生组织(WHO)于 2020 年 1 月 30 日宣布其为国际关注的突发公共卫生事件。本研究通过分子对接和分子动力学模拟研究,描述了如何将现有候选药物用作 SARS-CoV-2 M 的潜在抑制剂。通过使用从 Selleckchem Inc.(德克萨斯州休斯顿,http://www.selleckchem.com)和 DrugBank 数据库分别检索到的抗病毒和 FDA 批准药物库,应用药物再利用策略。使用 LeadIT FlexX 软件进行分子对接计算。进行了 100ns 的分子动力学模拟,以研究所有复合物系统的构象稳定性。来自 FDA 批准药物库的米托蒽醌和亚叶酸以及来自抗病毒药物库的 Birinapant 和 Dynasore 与 SARS-CoV-2 M 相互作用的效率更高,因为结合腔中的空间和疏水环境得到改善,从而形成稳定的复合物。此外,100ns 的分子动力学模拟揭示了所有复合物系统的平均 RMSD 值为 2.25Å。这表明先导化合物紧密结合在 M 腔中,因此具有构象稳定性。M 中的谷氨酸(Glu166)是通过形成氢键和盐桥来保持和形成报告的先导化合物稳定复合物的关键残基。我们的研究结果表明,米托蒽醌、亚叶酸、Birinapant 和 Dynasore 可能是 SARS-CoV-2 M 的抑制剂。