Cardiovascular Medicine (A.S.H., A.D.T., N.H., S.K., J.R., J.M.Y., H.W., S.S.), University of Michigan, Ann Arbor.
Molecular & Integrative Physiology (A.A.G.), University of Michigan, Ann Arbor.
Circ Genom Precis Med. 2020 Oct;13(5):396-405. doi: 10.1161/CIRCGEN.120.002929. Epub 2020 Aug 25.
Pathogenic variants in , encoding cardiac MyBP-C (myosin binding protein C), are the most common cause of familial hypertrophic cardiomyopathy. A large number of unique variants and relatively small genotyped hypertrophic cardiomyopathy cohorts have precluded detailed genotype-phenotype correlations.
Patients with hypertrophic cardiomyopathy and variants were identified from the Sarcomeric Human Cardiomyopathy Registry. Variant types and locations were analyzed, morphological severity was assessed, and time-event analysis was performed (composite clinical outcome of sudden death, class III/IV heart failure, left ventricular assist device/transplant, atrial fibrillation). For selected missense variants falling in enriched domains, myofilament localization and degradation rates were measured in vitro.
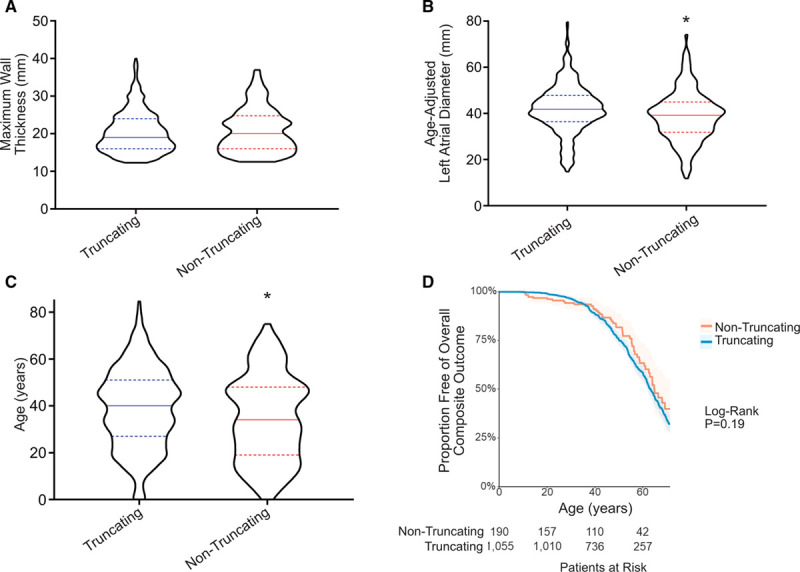
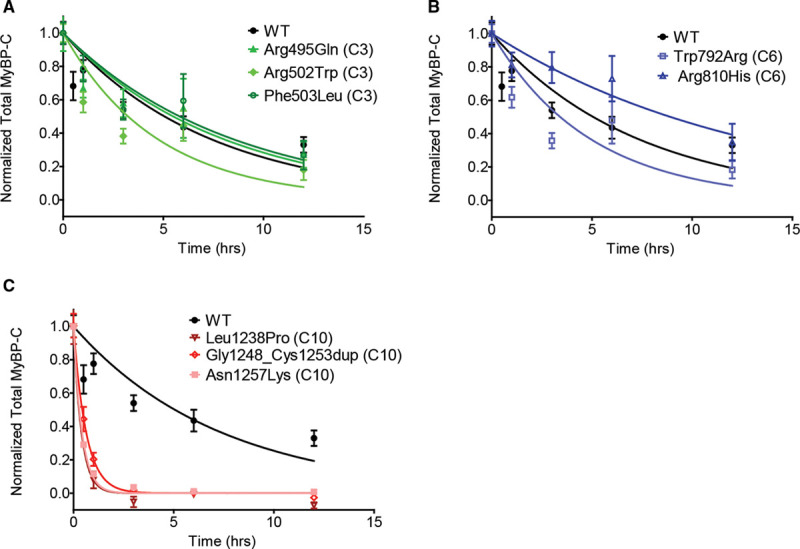
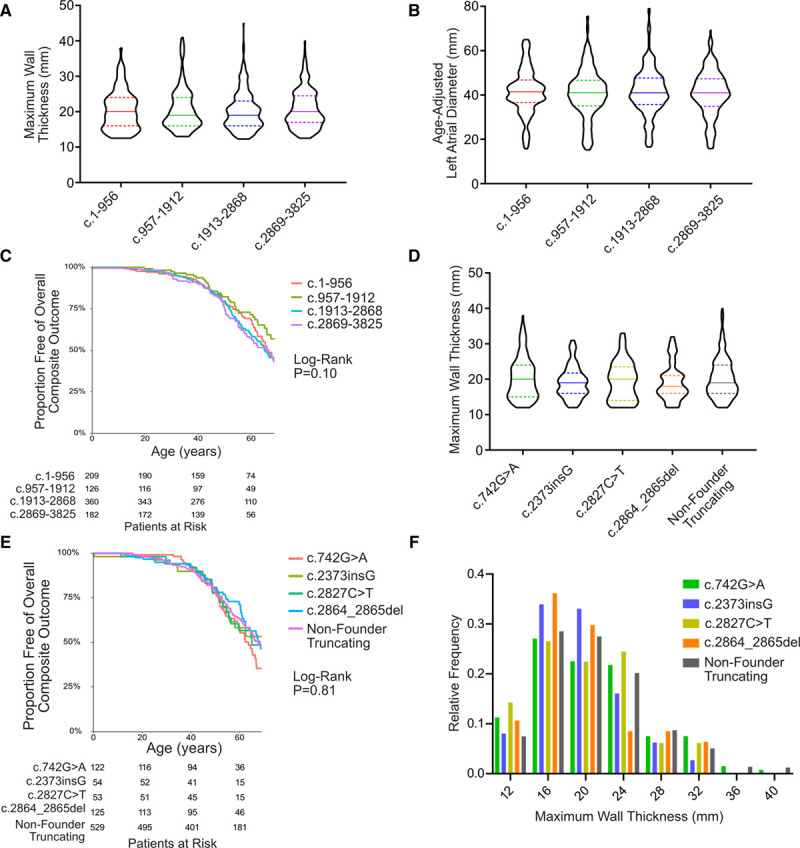
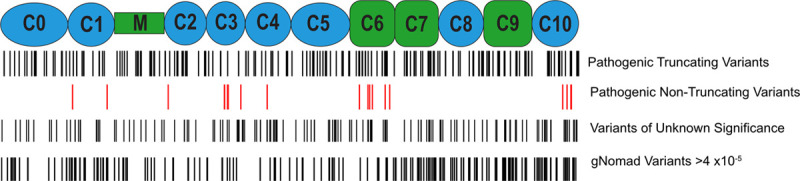
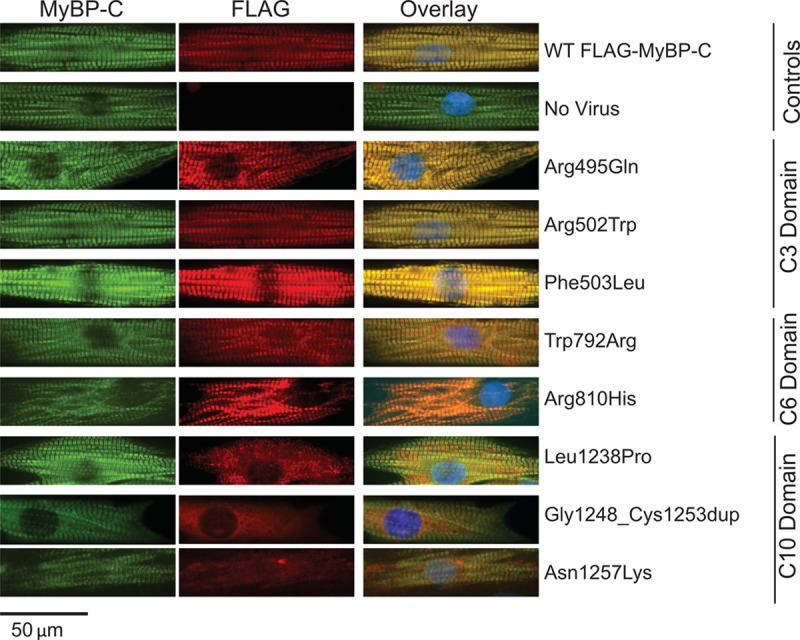
Among 4756 genotyped patients with hypertrophic cardiomyopathy in Sarcomeric Human Cardiomyopathy Registry, 1316 patients were identified with adjudicated pathogenic truncating (N=234 unique variants, 1047 patients) or nontruncating (N=22 unique variants, 191 patients) variants in . Truncating variants were evenly dispersed throughout the gene, and hypertrophy severity and outcomes were not associated with variant location (grouped by 5'-3' quartiles or by founder variant subgroup). Nontruncating pathogenic variants clustered in the C3, C6, and C10 domains (18 of 22, 82%, <0.001 versus Genome Aggregation Database common variants) and were associated with similar hypertrophy severity and adverse event rates as observed with truncating variants. MyBP-C with variants in the C3, C6, and C10 domains was expressed in rat ventricular myocytes. C10 mutant MyBP-C failed to incorporate into myofilaments and degradation rates were accelerated by ≈90%, while C3 and C6 mutant MyBP-C incorporated normally with degradation rate similar to wild-type.
Truncating variants account for 91% of pathogenic variants and cause similar clinical severity and outcomes regardless of location, consistent with locus-independent loss-of-function. Nontruncating pathogenic variants are regionally clustered, and a subset also cause loss of function through failure of myofilament incorporation and rapid degradation. Cardiac morphology and clinical outcomes are similar in patients with truncating versus nontruncating variants.
编码心肌肌球蛋白结合蛋白 C(myosin binding protein C)的 基因中的致病性变异是家族性肥厚型心肌病的最常见原因。大量独特的 变异和相对较小的肥厚型心肌病基因型队列妨碍了详细的基因型-表型相关性研究。
从肌节性人类心肌病注册中心中确定患有肥厚型心肌病和 变异的患者。分析变异类型和位置,评估形态严重程度,并进行时间事件分析(复合临床结局为猝死、III/IV 级心力衰竭、左心室辅助装置/移植、心房颤动)。对于落入富集区域的选定错义变异,在体外测量肌丝定位和降解率。
在肌节性人类心肌病注册中心中,对 4756 名基因型肥厚型心肌病患者进行基因分型,确定了 1316 名患者存在经裁定的截断(N=234 种独特变异,1047 名患者)或非截断(N=22 种独特变异,191 名患者) 变异。截断变异均匀分布在整个基因中,并且变异位置与肥厚严重程度和结局无关(按 5'-3'四分位数或创始变异亚组分组)。非截断致病性变异聚集在 C3、C6 和 C10 结构域(22 个中的 18 个,82%,<0.001 与基因组聚集数据库常见变异相比),并且与截断变异观察到的类似肥厚严重程度和不良事件发生率相关。在大鼠心室肌细胞中表达具有 C3、C6 和 C10 结构域变异的 MyBP-C。C10 突变型 MyBP-C 未能整合到肌丝中,降解率加快约 90%,而 C3 和 C6 突变型 MyBP-C 正常整合,降解率与野生型相似。
截断变异占 致病性变异的 91%,无论位置如何,都会导致相似的临床严重程度和结局,这与位置无关,符合无功能丧失的功能丧失。非截断 致病性变异呈区域性聚集,其中一部分也因肌丝整合失败和快速降解而导致功能丧失。在截断与非截断变异患者中,心脏形态和临床结局相似。