Panagiotopoulos Anna-Lena, Karguth Nina, Pavlou Marina, Böhm Sybille, Gasparoni Gilles, Walter Jörn, Graf Alexander, Blum Helmut, Biel Martin, Riedmayr Lisa Maria, Becirovic Elvir
Department of Pharmacy, Center for Drug Research, Ludwig-Maximilians-Universität München, Munich, Germany.
Center for Integrated Protein Science Munich CIPSM, Munich, Germany; Department of Pharmacy, Center for Drug Research, Ludwig-Maximilians-Universität München, Munich, Germany.
Mol Ther Nucleic Acids. 2020 Sep 4;21:1050-1061. doi: 10.1016/j.omtn.2020.07.036. Epub 2020 Jul 31.
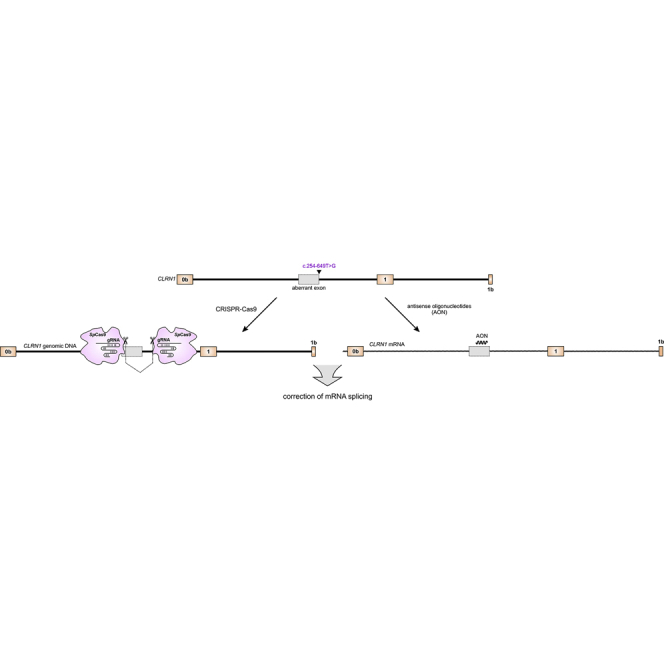
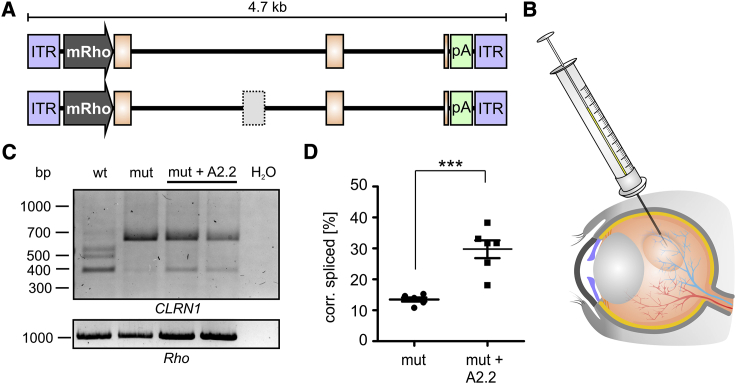
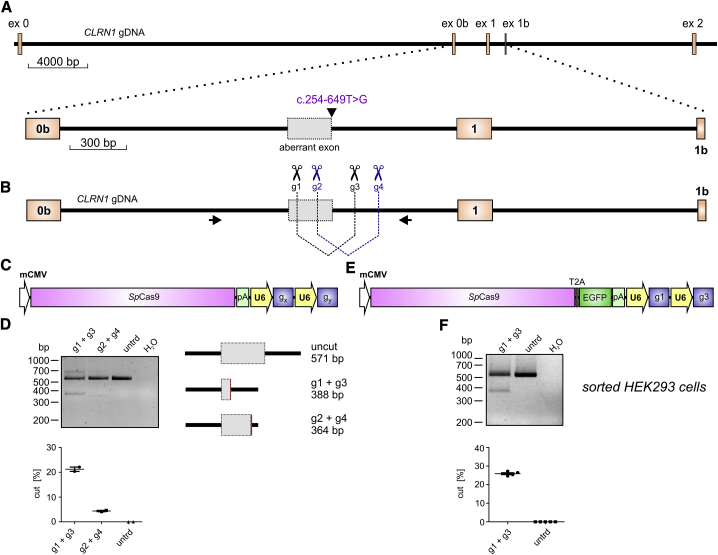
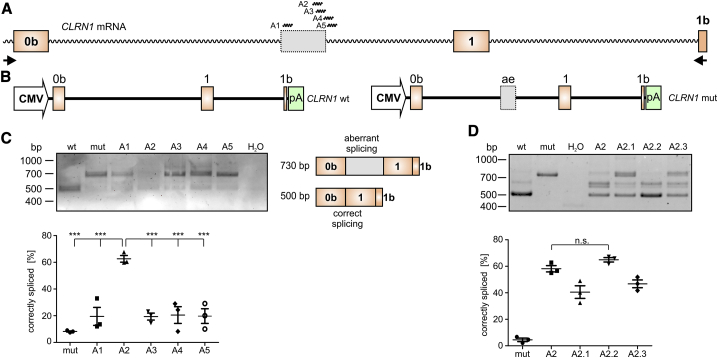
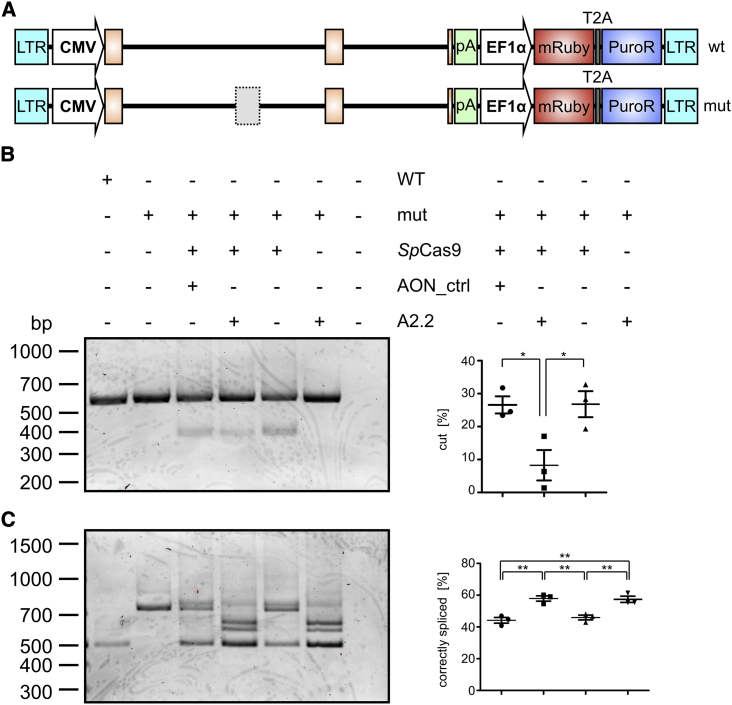
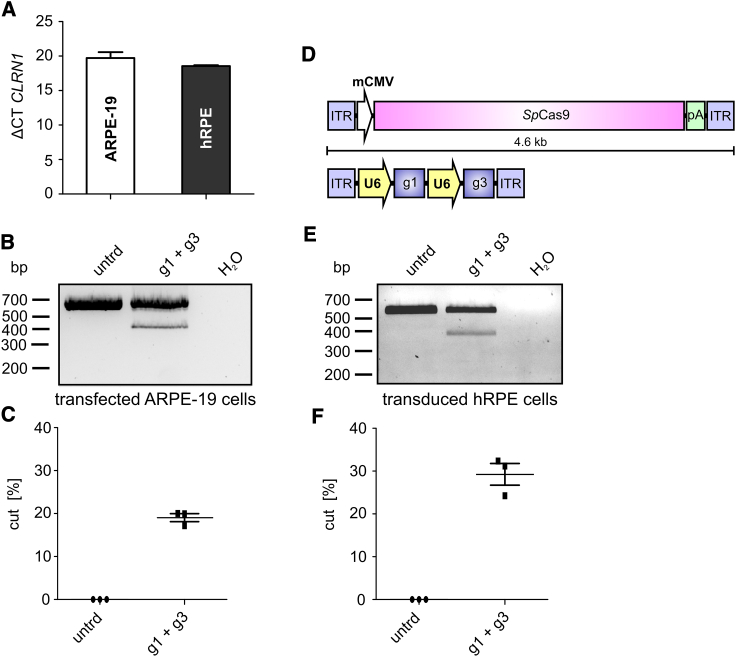
Mutations in CLRN1 cause Usher syndrome (USH) type III (USH3A), a disease characterized by progressive hearing impairment, retinitis pigmentosa, and vestibular dysfunction. Due to the lack of appropriate disease models, no efficient therapy for retinitis pigmentosa in USH patients exists so far. In addition, given the yet undefined functional role and expression of the different CLRN1 splice isoforms in the retina, non-causative therapies such as gene supplementation are unsuitable at this stage. In this study, we focused on the recently identified deep intronic c.254-649T>G CLRN1 splicing mutation and aimed to establish two causative treatment approaches: CRISPR-Cas9-mediated excision of the mutated intronic region and antisense oligonucleotide (AON)-mediated correction of mRNA splicing. The therapeutic potential of these approaches was validated in different cell types transiently or stably expressing CLRN1 minigenes. Both approaches led to substantial correction of the splice defect. Surprisingly, however, no synergistic effect was detected when combining both methods. Finally, the injection of naked AONs into mice expressing the mutant CLRN1 minigene in the retina also led to a significant splice rescue. We propose that both AONs and CRISPR-Cas9 are suitable strategies to initiate advanced preclinical studies for treatment of USH3A patients.
CLRN1基因的突变会导致III型遗传性耳聋-视网膜色素变性综合征(USH3A),该疾病的特征是进行性听力障碍、视网膜色素变性和前庭功能障碍。由于缺乏合适的疾病模型,目前尚无针对USH患者视网膜色素变性的有效治疗方法。此外,鉴于CLRN1不同剪接异构体在视网膜中的功能作用和表达尚未明确,基因补充等非病因性治疗方法在现阶段并不适用。在本研究中,我们聚焦于最近发现的CLRN1基因内含子深处的c.254-649T>G剪接突变,并旨在建立两种病因性治疗方法:CRISPR-Cas9介导的突变内含子区域切除和反义寡核苷酸(AON)介导的mRNA剪接校正。这些方法的治疗潜力在瞬时或稳定表达CLRN1小基因的不同细胞类型中得到了验证。两种方法均导致剪接缺陷得到显著校正。然而,令人惊讶的是,将两种方法联合使用时未检测到协同效应。最后,将裸露的AON注射到视网膜中表达突变CLRN1小基因的小鼠体内也导致了显著的剪接挽救。我们认为,AON和CRISPR-Cas9都是启动针对USH3A患者治疗的高级临床前研究的合适策略。